Genetic engineering from A to Z part 2
So, it’s time to continue the article on how to make a glowing Christmas tree next year with the use of real genetic engineering, and not the one about which you could read in the news :)
Summary of the previous series :
Scientists have discovered the gene blue glow. We read about this gene and caught fire to make a glowing transgenic tree. We found its name and sequence in the specialized resources, knocked out a business trip from the boss and went to where the animal lives - the buttery containing the gene.
By various tricks with the use of special equipment, we obtained pure bl1 DNA molecules encoding a protein of blue luminescence.
')
We have a gene. What are we waiting for, readers will ask, let's put this gene in the Christmas tree and it will start to glow?
Not everything is so simple, and here, why.
Any gene works only when information is read from it. In our case, this is mRNA of the bl1 protein. Moreover, the mRNA itself should work, but this is a separate story.
The second is that a gene must be transmitted when a cell divides to its descendants. Otherwise, everything will go down the drain.
The third is that the gene is not a pin, you still need to shove it!
If we just put a gene in a cell, then neither the information will be read from it, nor will it be transmitted (yes, actually, with its poking, it’s a big problem). Therefore, we must hang on him any official information that would allow him to work and clothe in a form that will allow him to pass on to his descendants. And also somehow put it in a cage.
However, even earlier we will try to preserve the existing gene, so that at any moment you can get a lot of it and not go for a boot.
The first transformation.
The very first task that stands at each stage is to preserve the results of the previous stages. Actual challenges in programming, is not it? :)
Now we will place our gene in a bacterium, so that at any moment we can multiply it and get the necessary quantity from there. This will be our first transgenic organism and the process of its creation is called transformation.
Why we just can not reproduce the gene using PCR, as we did before? The fact is that the probability of error in the reaction of PCR is 1/10 ^ 3, and in bacteria it is 1/10 ^ 6, that is, it is copied a thousand times more accurately, and we need absolute accuracy and the more, the better.
So, we put the gene in the bacterium, how to do it? To do this, first prepare a container - a special circular DNA molecule, called a plasmid . This plasmid can float in the bacterial cell, regardless of its genome, to multiply and be transmitted to descendants, since it has the necessary genes and signal sequences for this.
Also in the plasmid there are marker genes. These are genes that will help us select cells with a plasmid, from cells where it does not exist. A marker, for example, can be a gene for staining (bacteria with a plasmid will be colored) or resistance (bacteria with a plasmid will not die on a medium with an antibiotic).
Plasmids can be bought at a biotechnology company, they are sold to everyone. It will come to you as a clear solution in a small test tube (in fact, the solution of any other DNA also looks like). To insert a gene into a plasmid, it must be incised with an enzyme, a restriction enzyme (we’ll talk about it later), but often they are sold already incised.
Suppose we bought a modern plasmid, for example pJET1,2 from Fermentas .

Here it is, beautiful. In the upper right corner are many small names - this lists restriction sites. Rep is the site responsible for the replication (reproduction) of the plasmid in the cell, and Amp is the reporter gene for resistance to the antibiotic, ampicillin.
A kit is supplied to the plasmid, the so-called “kit”. We drop a little bit of the solution of our gene, a plasmid with reagents from the whale and an enzyme ligase, according to the instructions, and we get a solution where the plasmid molecules fused with the gene molecules and we got a single ring molecule containing the bl1 gene.
This plasmid is called an “insert plasmid”.
Enzyme ligase stitches docked to each other stretches of DNA. It stitches everything and cannot determine whether everything is properly docked (for example, it can simply stitch the plasmid to itself, without our gene at all), so our task is to make the molecules dock only in a certain way. A little further I will talk about this when it comes to sticky ends.
Plasmid pJet1,2 is sold already cut in the middle of the eco471R gene. This gene encodes cellular poison - a powerful restriction enzyme that will kill a cell if a ligase makes a plasmid without an inserted bl1 gene and such a plasmid will enter the bacterium. This is very convenient, since we do not need to worry that bacteria will grow in which there is no insert at all, which happens with other types of plasmids (this type is called a suicide plasmid or “selfkill plasmid”).
Initially, this information was beyond the scope of the article, but I thought that otherwise there would be questions why this gene :)
Next, take a specially prepared culture of Escherichia coli bacteria (Echerichia coli or E.coli) and transfer the plasmid solution with the insert according to the transformation protocol. The protocols may be different, here is an example . Bacteria are very easy to transform, in fact, DNA simply penetrates into them and all, no other tricks.
Usually the transformation is done in the evening, the next morning the bacteria grow up and we already have the results.
On a Petri dish with medium for growth, there are many colonies. Each colony is the descendants of a single transformed cell. We select some of them, test for bl1 content by PCR (to make sure the gene is present), and we can put it in storage.
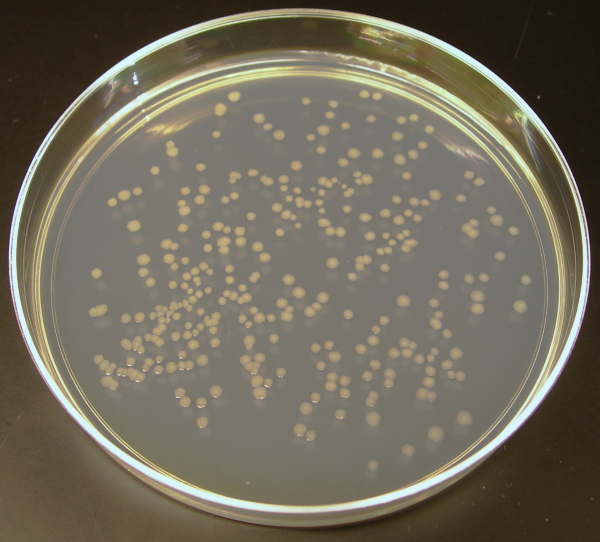
Bacteria grow very quickly and now the plasmid with the gene can be isolated from them at any time in the right quantity
So, we sowed :) and in parallel received a culture of transgenic bacteria. Whether they will glow or not depends on which plasmid for transformation we have chosen, whether it contains a signal sequence for the synthesis of an mRNA - a promoter.
I note that here such preservation of intermediate results is always carried out in bacteria, regardless of which object the work happens next. It is just very convenient to work and store with them (by quickly freezing in liquid nitrogen).
Preparation of transcribed sequence.
For a gene sequence to work in a cell, mRNA must be read from it (and protein from it), that is, it must be transcribed. In order for this to happen, there must necessarily be a control sequence, a promoter , in front of the gene.
All genes have promoters and they are responsible for the conditions under which the gene is to be synthesized, but certain promoters work only in certain organisms. Thus, bacterial promoters do not work in plants and vice versa, plant promoters do not work in bacteria. Since we want the tree to shine brightly, we will put the gene under the super-promoter, which works constantly. For plants, it may be the 35S promoter from cauliflower mosaic virus.
Fortunately, you don’t need to search for this virus and cauliflower :) You can buy a ready-made plasmid containing a promoter and having a place to insert a gene. For example, such as in the figure.

The next step is to isolate a plasmid with an insert from a culture of bacteria and cut out a gene from there. Cutting is performed using restriction enzymes (just add the enzyme and the buffer for the enzyme to the plasmid DNA solution). On the sides of the gene we have inserted, the plasmid contains DNA segments (restriction sites), which they recognize and cut.
You should always choose those restricts that will cut only in one place, without cutting the gene itself (a certain restriction enzyme always recognizes a certain site, which usually consists of 6 specific nucleotides). For example, the restriction enzyme EcoR1 will always cut the DNA if it detects the sequence G'AATTC in it, and one strand and the second strand will be cut.

If a restriction enzyme cuts not exactly in the middle of the recognition sequence, then the so-called “sticky ends” remain on the edges, where a part of the DNA is single-stranded. Such ends tend to “stick” - dock to other sticky ends with the same sequence of single-stranded segments. If the sequence is different, the docking will not occur. Thus, if we cut out a gene with two restrictases, one for the beginning of the gene and the other for the end, then its beginning and end will have different ends that will connect only with their own kind. This is very important for controlling the orientation of the gene, because it works only if it is rotated in a certain way to the service sequences.
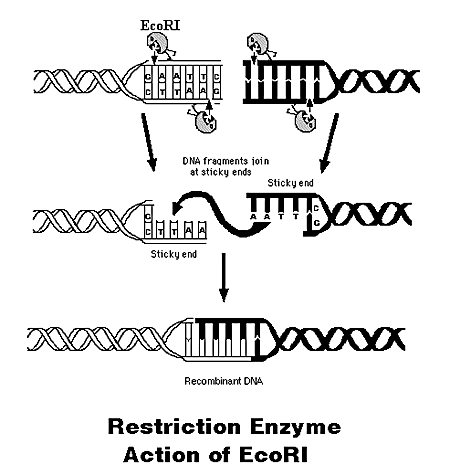
Some restricts can cut clearly in the middle and get “blunt ends”. On blunt ends, you can also make a stitching, but you’ll get a 50% chance of stitching a gene with the wrong side, but backwards.
After cutting out the gene, we clear it of plasmid residues by electrophoresis.
Similarly, we cut the purchased plasmid with a promoter and mix it with the excised gene.
We add ligases and voila - we have a stitched sequence, where we have the 35S promoter and the bl1 gene.
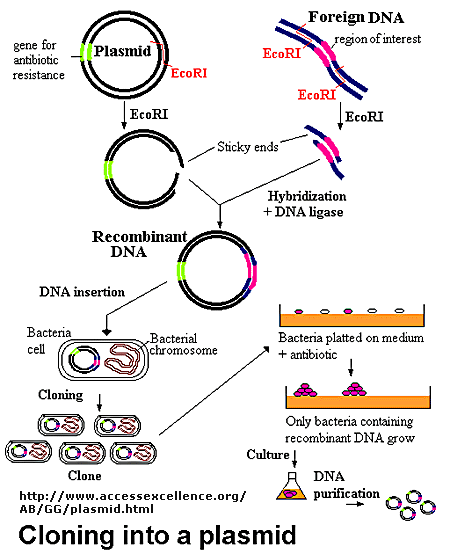
Again we transform bacteria, already by this obtained construction. Since we have introduced a promoter that works on plants, such bacteria will not glow. If we introduced a bacterial promoter, then in this step the culture of bacteria would be lit.
A plasmid with a promoter, a gene, markers and other auxiliary parts is called “expression plasmid”. It can already be used to produce transgenic plants.
This is our second save. We already know how to make bacteria glow and got almost everything needed to get transgenic plants.
Note that the process of genetic engineering is multistage and multitasking. In real life, there is a simultaneous work with 3-4 experiments at different stages. While bacteria grow in one, for the other we are restricting a plasmid, and for the third we isolate a gene. This happens due to the fact that never so perfectly smooth course of work, as I described, problems always arise for different reasons, they have to be discovered, overcome. Even finding an error at each stage takes at least several hours.
And it is to overcome the problems that a whole amount of knowledge of a molecular biologist is needed, and not for the sequential dripping from one tube to another, which a laboratory technician can do :)
But about how we still get a glowing blue Christmas tree, I will tell in the final article. In the same place I will provide a list of equipment and reagents, with prices :)
UPD:
Part 3 , the final.
Summary of the previous series :
Scientists have discovered the gene blue glow. We read about this gene and caught fire to make a glowing transgenic tree. We found its name and sequence in the specialized resources, knocked out a business trip from the boss and went to where the animal lives - the buttery containing the gene.
By various tricks with the use of special equipment, we obtained pure bl1 DNA molecules encoding a protein of blue luminescence.
')
We have a gene. What are we waiting for, readers will ask, let's put this gene in the Christmas tree and it will start to glow?
Not everything is so simple, and here, why.
Any gene works only when information is read from it. In our case, this is mRNA of the bl1 protein. Moreover, the mRNA itself should work, but this is a separate story.
The second is that a gene must be transmitted when a cell divides to its descendants. Otherwise, everything will go down the drain.
The third is that the gene is not a pin, you still need to shove it!
If we just put a gene in a cell, then neither the information will be read from it, nor will it be transmitted (yes, actually, with its poking, it’s a big problem). Therefore, we must hang on him any official information that would allow him to work and clothe in a form that will allow him to pass on to his descendants. And also somehow put it in a cage.
However, even earlier we will try to preserve the existing gene, so that at any moment you can get a lot of it and not go for a boot.
The first transformation.
The very first task that stands at each stage is to preserve the results of the previous stages. Actual challenges in programming, is not it? :)
Now we will place our gene in a bacterium, so that at any moment we can multiply it and get the necessary quantity from there. This will be our first transgenic organism and the process of its creation is called transformation.
Why we just can not reproduce the gene using PCR, as we did before? The fact is that the probability of error in the reaction of PCR is 1/10 ^ 3, and in bacteria it is 1/10 ^ 6, that is, it is copied a thousand times more accurately, and we need absolute accuracy and the more, the better.
So, we put the gene in the bacterium, how to do it? To do this, first prepare a container - a special circular DNA molecule, called a plasmid . This plasmid can float in the bacterial cell, regardless of its genome, to multiply and be transmitted to descendants, since it has the necessary genes and signal sequences for this.
Also in the plasmid there are marker genes. These are genes that will help us select cells with a plasmid, from cells where it does not exist. A marker, for example, can be a gene for staining (bacteria with a plasmid will be colored) or resistance (bacteria with a plasmid will not die on a medium with an antibiotic).
Plasmids can be bought at a biotechnology company, they are sold to everyone. It will come to you as a clear solution in a small test tube (in fact, the solution of any other DNA also looks like). To insert a gene into a plasmid, it must be incised with an enzyme, a restriction enzyme (we’ll talk about it later), but often they are sold already incised.
Suppose we bought a modern plasmid, for example pJET1,2 from Fermentas .
Here it is, beautiful. In the upper right corner are many small names - this lists restriction sites. Rep is the site responsible for the replication (reproduction) of the plasmid in the cell, and Amp is the reporter gene for resistance to the antibiotic, ampicillin.
A kit is supplied to the plasmid, the so-called “kit”. We drop a little bit of the solution of our gene, a plasmid with reagents from the whale and an enzyme ligase, according to the instructions, and we get a solution where the plasmid molecules fused with the gene molecules and we got a single ring molecule containing the bl1 gene.
This plasmid is called an “insert plasmid”.
Enzyme ligase stitches docked to each other stretches of DNA. It stitches everything and cannot determine whether everything is properly docked (for example, it can simply stitch the plasmid to itself, without our gene at all), so our task is to make the molecules dock only in a certain way. A little further I will talk about this when it comes to sticky ends.
Plasmid pJet1,2 is sold already cut in the middle of the eco471R gene. This gene encodes cellular poison - a powerful restriction enzyme that will kill a cell if a ligase makes a plasmid without an inserted bl1 gene and such a plasmid will enter the bacterium. This is very convenient, since we do not need to worry that bacteria will grow in which there is no insert at all, which happens with other types of plasmids (this type is called a suicide plasmid or “selfkill plasmid”).
Initially, this information was beyond the scope of the article, but I thought that otherwise there would be questions why this gene :)
Next, take a specially prepared culture of Escherichia coli bacteria (Echerichia coli or E.coli) and transfer the plasmid solution with the insert according to the transformation protocol. The protocols may be different, here is an example . Bacteria are very easy to transform, in fact, DNA simply penetrates into them and all, no other tricks.
Usually the transformation is done in the evening, the next morning the bacteria grow up and we already have the results.
On a Petri dish with medium for growth, there are many colonies. Each colony is the descendants of a single transformed cell. We select some of them, test for bl1 content by PCR (to make sure the gene is present), and we can put it in storage.
Bacteria grow very quickly and now the plasmid with the gene can be isolated from them at any time in the right quantity
So, we sowed :) and in parallel received a culture of transgenic bacteria. Whether they will glow or not depends on which plasmid for transformation we have chosen, whether it contains a signal sequence for the synthesis of an mRNA - a promoter.
I note that here such preservation of intermediate results is always carried out in bacteria, regardless of which object the work happens next. It is just very convenient to work and store with them (by quickly freezing in liquid nitrogen).
Preparation of transcribed sequence.
For a gene sequence to work in a cell, mRNA must be read from it (and protein from it), that is, it must be transcribed. In order for this to happen, there must necessarily be a control sequence, a promoter , in front of the gene.
All genes have promoters and they are responsible for the conditions under which the gene is to be synthesized, but certain promoters work only in certain organisms. Thus, bacterial promoters do not work in plants and vice versa, plant promoters do not work in bacteria. Since we want the tree to shine brightly, we will put the gene under the super-promoter, which works constantly. For plants, it may be the 35S promoter from cauliflower mosaic virus.
Fortunately, you don’t need to search for this virus and cauliflower :) You can buy a ready-made plasmid containing a promoter and having a place to insert a gene. For example, such as in the figure.
The next step is to isolate a plasmid with an insert from a culture of bacteria and cut out a gene from there. Cutting is performed using restriction enzymes (just add the enzyme and the buffer for the enzyme to the plasmid DNA solution). On the sides of the gene we have inserted, the plasmid contains DNA segments (restriction sites), which they recognize and cut.
You should always choose those restricts that will cut only in one place, without cutting the gene itself (a certain restriction enzyme always recognizes a certain site, which usually consists of 6 specific nucleotides). For example, the restriction enzyme EcoR1 will always cut the DNA if it detects the sequence G'AATTC in it, and one strand and the second strand will be cut.
If a restriction enzyme cuts not exactly in the middle of the recognition sequence, then the so-called “sticky ends” remain on the edges, where a part of the DNA is single-stranded. Such ends tend to “stick” - dock to other sticky ends with the same sequence of single-stranded segments. If the sequence is different, the docking will not occur. Thus, if we cut out a gene with two restrictases, one for the beginning of the gene and the other for the end, then its beginning and end will have different ends that will connect only with their own kind. This is very important for controlling the orientation of the gene, because it works only if it is rotated in a certain way to the service sequences.
Some restricts can cut clearly in the middle and get “blunt ends”. On blunt ends, you can also make a stitching, but you’ll get a 50% chance of stitching a gene with the wrong side, but backwards.
After cutting out the gene, we clear it of plasmid residues by electrophoresis.
Similarly, we cut the purchased plasmid with a promoter and mix it with the excised gene.
We add ligases and voila - we have a stitched sequence, where we have the 35S promoter and the bl1 gene.
Again we transform bacteria, already by this obtained construction. Since we have introduced a promoter that works on plants, such bacteria will not glow. If we introduced a bacterial promoter, then in this step the culture of bacteria would be lit.
A plasmid with a promoter, a gene, markers and other auxiliary parts is called “expression plasmid”. It can already be used to produce transgenic plants.
This is our second save. We already know how to make bacteria glow and got almost everything needed to get transgenic plants.
Note that the process of genetic engineering is multistage and multitasking. In real life, there is a simultaneous work with 3-4 experiments at different stages. While bacteria grow in one, for the other we are restricting a plasmid, and for the third we isolate a gene. This happens due to the fact that never so perfectly smooth course of work, as I described, problems always arise for different reasons, they have to be discovered, overcome. Even finding an error at each stage takes at least several hours.
And it is to overcome the problems that a whole amount of knowledge of a molecular biologist is needed, and not for the sequential dripping from one tube to another, which a laboratory technician can do :)
But about how we still get a glowing blue Christmas tree, I will tell in the final article. In the same place I will provide a list of equipment and reagents, with prices :)
UPD:
Part 3 , the final.
Source: https://habr.com/ru/post/48846/
All Articles