Genetic engineering from A to Z
Greetings dear community!
So, this is my first post on Habré :)
It will be devoted to a serious topic in which, by the will of fate, I am well versed. Namely, genetic engineering.
I remember that here I ran through a post in which it was said about the gennotechnological laboratory “on the knee”. It turned out that the topic is interesting to the audience, so I decided to take up its development with educational goals.
')
I will give clear and understandable examples for ordinary people to describe complex processes. If someone considers it necessary to correct me - do not hesitate. I will deliberately miss many things, but if it seems to you that without them the logic of the presentation suffers - just correct.
So, let's begin. Suppose we want to create a transgenic Christmas tree glowing blue light. For example, British scientists just recently discovered the gene of the blue glow. So let's take a look at this process in stages.
Gene glow.
We will conduct an experiment like real scientists. They hear that a new gene is discovered, what should they do next if they want to create a Christmas tree?
A real scientist usually climbs into ncbi.nih.gov and for several key words is looking for scientific publications on this topic. For example, the “blue glow gene shines”. A typical situation is when, according to one of the links, he actually finds an article by “British scientists”, which turns out to be an article by a group of Chinese authors, none of whom respond to an e-mail.
On the other hand, in the article you can find out the name of this gene. Let it be called ButiBl1 (the names of the genes are usually given by letter + index, and the first few letters of the name of the organism from which it is selected can go in front of them, you can discard them). ButiBl1 , for example, can be decoded as Butiavka marina blue light 1 gene. But the rules are not strict.
According to the name of the gene in the database of nucleotides are looking for the gene sequence. This is what the scientist sees about the screen.
By the way, we can use the BLAST tool and enter the DNA sequence, get what genes it can belong to. This is also a very important routine tool for genetic engineers.
So we got the gene sequence. Very good, what next? You need to get the gene itself. To do this, let us return to the question of what DNA is.
About DNA.
DNA is a long molecule (very long), is a polymer of four variants of small molecules - nitrogenous bases, simply “letters”.
The cell's genome is divided into parts - from one to several dozen DNA molecules, and usually each of them also has its own twin-copy, carrying the same genes. Each of the DNA molecules is folded in a special way to fit in the cell and covered with protein complexes, forming the chromosome.
I hope everyone remembers this, but if you need to refresh your memory, please, in the wiki :)
So, remember the main thing:
1. DNA is a molecule.
2. Since this is a molecule, it is not visible through a microscope, do not pick it up with tweezers, etc. etc.
3. A number of DNA molecules are read in a cell, and if there are many of them, they are heterogeneous and “pick them up with a bunch” to pick them up with tweezers (point 2) also fail.
How do genetic engineers work with a DNA molecule if it is alone and it is impossible to carry out any direct manipulations with it? The fact is that in all procedures there is work not with one, but with a multitude of DNA molecules, with thousands and millions of its copies.
Thousands of such identical molecules float in an aqueous solution and this solution is called a “DNA preparation”. All molecule manipulations are carried out using typical chemical methods.
That is, scientists work not with a single molecule, but with a huge amount of them in solution using chemical methods.
How do we get the bl1 gene? There are two ways. The first is direct chemical synthesis. However, they do not get sufficiently long molecules due to synthesis errors. I will explain why.
DNA is a polymer. It can be synthesized by building up brick by brick, and there are four bricks of different colors. At each stage of building efficiency is about 99%. That is, out of a hundred molecules one turns out to be wrong. Now imagine that we need to make a molecule with a length of 1000 letters? Then applying banal arithmetic it turns out that the proportion of true molecules will be 0.99 ^ 1000 = 0.00004
Considering that it is almost impossible to separate right and wrong molecules, our undertaking will fail here, and in real problems the synthesis of more than 100-150 letters already seems to be unrealistic.
There is a second way.
Goodwill
We beat a business trip out of our boss on the coast of the Maldives, where the notorious butiavka marina is found.
We catch it, pound it into powder, fill it with successively different chemical filthies so that only DNA molecules remain in the solution from the whole mass of tissues. The end result of this is the butyavki DNA preparation. Since the selection is made from a relatively large sample, there is not one DNA molecule, but a lot - from each cell for a pair of pieces. This DNA contains not only the bl1 gene, but all the other butyavo genes.
This step is called DNA extraction. It can be isolated not only in the form of a solution, but it can also be transplanted and a dry preparation, that is, pure DNA molecules.
Amplification
So, the trip is over, so we will rush back to the laboratory where we are waiting for a wonderful amplification procedure.
See, in the DNA preparation of a butterfly, a bunch of all sorts of different genes, and not just the one we need. We can only work with homogeneous preparations, we need to bring the content of the DNA molecules of the bl1 gene to at least 90 percent.
And here we use a truly wonderful technique, which is the cornerstone of modern bioengineering, called polymerase chain reaction or PCR (polymerase chain reaction, PCR). For the discovery of this method, they awarded the Nobel Prize, although there is still debate about priority, so I don’t give names to anyone who is interested - read .
The principle of the polymerase chain reaction is rather complicated, the explanation I will give is very rude and only in order to have at least some idea, for a detailed one, welcome to the link above.
So, we need to multiply (amplify) the DNA of a particular gene. To do this, we open a page with the sequence of our gene and find its ends. We take 20-30 letters from the end and the same from the beginning and synthesize short DNA molecules by chemical synthesis (usually done by special companies)
That is, we have two new tubes. In one of them, there are many short 30-letter DNA sequences that are homologous to the beginning of a gene, and the second is the same, but for the end of the gene. These new molecules are called primers.
Now we start the reaction of PCR, and we will multiply the area between the two primers (between the initial and terminal). A PCR reaction is a biochemical cyclic reaction that requires a change in temperature. At one time, it was made in water baths, but now they use special devices - amplifiers (they are also PCR machines). Their structure is very simple, there are Peltier elements, there is a place for test tubes and electronic brains and a control panel are attached to all this.

That is, we returned to the lab with the DNA of the bottle-up. We ordered two primers - to the beginning and to the end of the gene. Then they took a clean test tube, put a little DNA there, a little bit of each primer, a polymerase (an enzyme that builds DNA), nucleotides for building DNA, and some salts for the enzyme to work properly, put it in an amplifier for a couple of hours. In the amplifier, the mixture was then heated, then cooled, and at the exit we received a tube in which a lot of DNA copies of the gene we needed floated.
However, the test tube is transparent, how to see that there is some kind of DNA, and what else is necessary?

DNA detection.
There are many ways to see DNA, but I will describe a classic, called gel electrophoresis.
In the laboratory, there is a small bath with electrodes, called poreznoy camera.
A melt of an electrophoretic gel is poured into this bath, which in essence is very similar to marmalade. But instead of sugar, there are additives of salts and a fluorescent dye - ethidium bromide. This substance is interesting because it is embedded in the DNA molecule and in this case it starts to glow in the ultraviolet.
After the gel hardens, we apply the DNA preparation to the well on it, where supposedly there should already be many copies of the bl1 gene and turn on the electric current. In another well we put a “weight marker” - a special preparation of DNA molecules, consisting in equal parts of molecules of length 100, 200, 300, etc. nucleotides.
DNA molecules are polar and move in an electric field; the longer they are, the more they cling to the gel structure and the slower they move. After a while we turn off the electricity and carry the gel under an ultraviolet lamp.
On the path where we put the weight marker, we see a bunch of strips. The furthest from the place of application of the sample corresponds to the shortest DNA, the closest one to the longest.
In the neighboring holes, the DNA runs at the same speed, so we compare their position on the adjacent tracks and can determine the relative size.
So, we found on the track where we put a sample of one luminous strip and its size judging by the adjacent weight marker is what we expected.
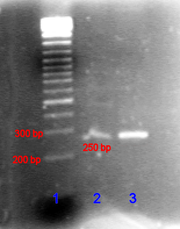
We carefully cut out the luminous piece of the blade from the gel - it contains a lot of bl1 DNA entangled in the gel and with the help of special manipulations we release molecules from it.
You can congratulate yourself, we have selected the bl1 gene from the booty!
I told only about the first stage of this complex and long process. Does it continue? You decide :)
upd. Part Two .
Part Three
upd2. Transferred to biotechnology
So, this is my first post on Habré :)
It will be devoted to a serious topic in which, by the will of fate, I am well versed. Namely, genetic engineering.
I remember that here I ran through a post in which it was said about the gennotechnological laboratory “on the knee”. It turned out that the topic is interesting to the audience, so I decided to take up its development with educational goals.
')
I will give clear and understandable examples for ordinary people to describe complex processes. If someone considers it necessary to correct me - do not hesitate. I will deliberately miss many things, but if it seems to you that without them the logic of the presentation suffers - just correct.
So, let's begin. Suppose we want to create a transgenic Christmas tree glowing blue light. For example, British scientists just recently discovered the gene of the blue glow. So let's take a look at this process in stages.
Gene glow.
We will conduct an experiment like real scientists. They hear that a new gene is discovered, what should they do next if they want to create a Christmas tree?
A real scientist usually climbs into ncbi.nih.gov and for several key words is looking for scientific publications on this topic. For example, the “blue glow gene shines”. A typical situation is when, according to one of the links, he actually finds an article by “British scientists”, which turns out to be an article by a group of Chinese authors, none of whom respond to an e-mail.
On the other hand, in the article you can find out the name of this gene. Let it be called ButiBl1 (the names of the genes are usually given by letter + index, and the first few letters of the name of the organism from which it is selected can go in front of them, you can discard them). ButiBl1 , for example, can be decoded as Butiavka marina blue light 1 gene. But the rules are not strict.
According to the name of the gene in the database of nucleotides are looking for the gene sequence. This is what the scientist sees about the screen.
By the way, we can use the BLAST tool and enter the DNA sequence, get what genes it can belong to. This is also a very important routine tool for genetic engineers.
So we got the gene sequence. Very good, what next? You need to get the gene itself. To do this, let us return to the question of what DNA is.
About DNA.
DNA is a long molecule (very long), is a polymer of four variants of small molecules - nitrogenous bases, simply “letters”.
The cell's genome is divided into parts - from one to several dozen DNA molecules, and usually each of them also has its own twin-copy, carrying the same genes. Each of the DNA molecules is folded in a special way to fit in the cell and covered with protein complexes, forming the chromosome.
I hope everyone remembers this, but if you need to refresh your memory, please, in the wiki :)
So, remember the main thing:
1. DNA is a molecule.
2. Since this is a molecule, it is not visible through a microscope, do not pick it up with tweezers, etc. etc.
3. A number of DNA molecules are read in a cell, and if there are many of them, they are heterogeneous and “pick them up with a bunch” to pick them up with tweezers (point 2) also fail.
How do genetic engineers work with a DNA molecule if it is alone and it is impossible to carry out any direct manipulations with it? The fact is that in all procedures there is work not with one, but with a multitude of DNA molecules, with thousands and millions of its copies.
Thousands of such identical molecules float in an aqueous solution and this solution is called a “DNA preparation”. All molecule manipulations are carried out using typical chemical methods.
That is, scientists work not with a single molecule, but with a huge amount of them in solution using chemical methods.
How do we get the bl1 gene? There are two ways. The first is direct chemical synthesis. However, they do not get sufficiently long molecules due to synthesis errors. I will explain why.
DNA is a polymer. It can be synthesized by building up brick by brick, and there are four bricks of different colors. At each stage of building efficiency is about 99%. That is, out of a hundred molecules one turns out to be wrong. Now imagine that we need to make a molecule with a length of 1000 letters? Then applying banal arithmetic it turns out that the proportion of true molecules will be 0.99 ^ 1000 = 0.00004
Considering that it is almost impossible to separate right and wrong molecules, our undertaking will fail here, and in real problems the synthesis of more than 100-150 letters already seems to be unrealistic.
There is a second way.
Goodwill
We beat a business trip out of our boss on the coast of the Maldives, where the notorious butiavka marina is found.
We catch it, pound it into powder, fill it with successively different chemical filthies so that only DNA molecules remain in the solution from the whole mass of tissues. The end result of this is the butyavki DNA preparation. Since the selection is made from a relatively large sample, there is not one DNA molecule, but a lot - from each cell for a pair of pieces. This DNA contains not only the bl1 gene, but all the other butyavo genes.
This step is called DNA extraction. It can be isolated not only in the form of a solution, but it can also be transplanted and a dry preparation, that is, pure DNA molecules.
Amplification
So, the trip is over, so we will rush back to the laboratory where we are waiting for a wonderful amplification procedure.
See, in the DNA preparation of a butterfly, a bunch of all sorts of different genes, and not just the one we need. We can only work with homogeneous preparations, we need to bring the content of the DNA molecules of the bl1 gene to at least 90 percent.
And here we use a truly wonderful technique, which is the cornerstone of modern bioengineering, called polymerase chain reaction or PCR (polymerase chain reaction, PCR). For the discovery of this method, they awarded the Nobel Prize, although there is still debate about priority, so I don’t give names to anyone who is interested - read .
The principle of the polymerase chain reaction is rather complicated, the explanation I will give is very rude and only in order to have at least some idea, for a detailed one, welcome to the link above.
So, we need to multiply (amplify) the DNA of a particular gene. To do this, we open a page with the sequence of our gene and find its ends. We take 20-30 letters from the end and the same from the beginning and synthesize short DNA molecules by chemical synthesis (usually done by special companies)
That is, we have two new tubes. In one of them, there are many short 30-letter DNA sequences that are homologous to the beginning of a gene, and the second is the same, but for the end of the gene. These new molecules are called primers.
Now we start the reaction of PCR, and we will multiply the area between the two primers (between the initial and terminal). A PCR reaction is a biochemical cyclic reaction that requires a change in temperature. At one time, it was made in water baths, but now they use special devices - amplifiers (they are also PCR machines). Their structure is very simple, there are Peltier elements, there is a place for test tubes and electronic brains and a control panel are attached to all this.
That is, we returned to the lab with the DNA of the bottle-up. We ordered two primers - to the beginning and to the end of the gene. Then they took a clean test tube, put a little DNA there, a little bit of each primer, a polymerase (an enzyme that builds DNA), nucleotides for building DNA, and some salts for the enzyme to work properly, put it in an amplifier for a couple of hours. In the amplifier, the mixture was then heated, then cooled, and at the exit we received a tube in which a lot of DNA copies of the gene we needed floated.
However, the test tube is transparent, how to see that there is some kind of DNA, and what else is necessary?
DNA detection.
There are many ways to see DNA, but I will describe a classic, called gel electrophoresis.
In the laboratory, there is a small bath with electrodes, called poreznoy camera.
A melt of an electrophoretic gel is poured into this bath, which in essence is very similar to marmalade. But instead of sugar, there are additives of salts and a fluorescent dye - ethidium bromide. This substance is interesting because it is embedded in the DNA molecule and in this case it starts to glow in the ultraviolet.
After the gel hardens, we apply the DNA preparation to the well on it, where supposedly there should already be many copies of the bl1 gene and turn on the electric current. In another well we put a “weight marker” - a special preparation of DNA molecules, consisting in equal parts of molecules of length 100, 200, 300, etc. nucleotides.
DNA molecules are polar and move in an electric field; the longer they are, the more they cling to the gel structure and the slower they move. After a while we turn off the electricity and carry the gel under an ultraviolet lamp.
On the path where we put the weight marker, we see a bunch of strips. The furthest from the place of application of the sample corresponds to the shortest DNA, the closest one to the longest.
In the neighboring holes, the DNA runs at the same speed, so we compare their position on the adjacent tracks and can determine the relative size.
So, we found on the track where we put a sample of one luminous strip and its size judging by the adjacent weight marker is what we expected.
We carefully cut out the luminous piece of the blade from the gel - it contains a lot of bl1 DNA entangled in the gel and with the help of special manipulations we release molecules from it.
You can congratulate yourself, we have selected the bl1 gene from the booty!
I told only about the first stage of this complex and long process. Does it continue? You decide :)
upd. Part Two .
Part Three
upd2. Transferred to biotechnology
Source: https://habr.com/ru/post/48533/
All Articles