Parsing 25TB with AWK and R

How to read this article : I apologize for the fact that the text was so long and chaotic. To save your time, I begin each chapter with the introduction of “What I have learned,” in which I state the essence of the chapter with one or two sentences.
“Just show the solution!” If you just want to see what I’ve come to, go to the chapter “I’m becoming more resourceful,” but I think it’s more interesting and useful to read about failures.
Recently, I was instructed to customize the processing of a large amount of the original DNA sequences (technically, this is an SNP chip). It was necessary to quickly obtain data on a given genetic location (called SNP) for subsequent modeling and other tasks. With the help of R and AWK I managed to clear and organize the data in a natural way, greatly speeding up the processing of requests. It was hard for me and required numerous iterations. This article will help you to avoid some of my mistakes and show you what I finally did.
First, some introductory notes.
')
Data
Our university center for the processing of genetic information provided us with data in the form of a TSV of 25 TB. I got them broken into 5 packages compressed with Gzip, each of which contained about 240 four-gigabyte files. Each row contained data for one SNP of one person. In total, data on ~ 2.5 million SNPs and ~ 60 thousand people were transmitted. In addition to the SNP information, there were numerous columns in the files with numbers reflecting various characteristics, such as reading intensity, frequency of different alleles, etc. There were about 30 columns with unique values.
purpose
As with any data management project, the most important thing was to determine how the data would be used. In this case, we will mostly select models and workflows for SNPs based on SNPs . That is, at the same time we will need data for only one SNP. I had to learn how to retrieve all the records related to one of the 2.5 million SNPs as easily as possible, faster and cheaper.
How not to do that
I quote a suitable cliche:
I couldn’t fail a thousand times, I just discovered a thousand ways to not parse a bunch of data in a format that is convenient for queries.
First try
What I learned : there is no cheap way to hit 25 TB at a time.
After listening to the Vanderbilt University subject “Advanced methods for processing big data,” I was sure that this was in the bag. Perhaps an hour or two will be spent on setting up a Hive server to run through all the data and report on the result. Since our data is stored in AWS S3, I used the Athena service, which allows you to apply Hive SQL queries to S3 data. No need to configure / raise the Hive-cluster, and even pay only for the data you are looking for.
After I showed Athena my data and its format, I ran a few tests with similar requests:
select * from intensityData limit 10; And quickly got well-structured results. Is done.
While we have not tried to use the data in the work ...
I was asked to pull out all the information on the SNP to test the model on it. I ran the query:
select * from intensityData where snp = 'rs123456'; ... and waited. After eight minutes and more than 4 TB of the requested data, I got the result. Athena charges for the amount of data found, at $ 5 per terabyte. So this single request cost $ 20 and eight minutes of waiting. To get rid of the model according to all data, it was necessary to wait 38 years and pay $ 50 million. Obviously, this did not suit us.
It was necessary to use Parquet ...
What I learned : be careful with the size of your Parquet files and their organization.
At first, I tried to rectify the situation by converting all the TSVs into parquet files . They are convenient for working with large data sets, because the information in them is stored in a column view: each column lies in its own memory / disk segment, unlike text files, in which the lines contain elements of each column. And if you need to find something, it is enough to read the necessary column. In addition, each file in the column stores a range of values, so if the desired value is not in the range of the column, Spark will not waste time scanning the entire file.
I launched AWS Glue's simple task to convert our TSV to Parquet and threw new files into Athena. It took about 5 hours. But when I launched the request, it took about the same amount of time and a little less money. The fact is that Spark, trying to optimize the task, simply unpacked one TSV chunk and put it into its own Parquet chunk. And since each chunk was large enough to hold the full records of many people, every SNP was stored in each file, so Spark had to open all the files to extract the necessary information.
Curiously, the default (and recommended) type of compression in Parquet — snappy — is not splitable. Therefore, each executor stuck on the unpacking task and download a full 3.5 GB dataset.

We understand the problem
What I learned : sorting is difficult, especially if the data is distributed.
It seemed to me that now I understood the essence of the problem. I only needed to sort the data by SNP column, not by people. Then several SNPs will be stored in a separate data chunk, and then the clever Parquet function “open only if the value is in the range” will manifest itself in all its glory. Unfortunately, sorting the billions of lines scattered across a cluster has proven to be a challenge.
AWS doesn't exactly want to return the money due to “I am a scattered student.” After I started sorting on Amazon Glue, it worked for 2 days and ended in a crash.
What about partitioning?
What I learned : Spark partitions should be balanced.
Then I got the idea to partition the data into chromosomes. There are 23 of them (and a few more, if we take into account mitochondrial DNA and unscrambled regions).
This will divide the data into smaller portions. If you add to the Spark-export function in the Glue script just one line
partition_by = "chr" , then the data should be laid out in buckets.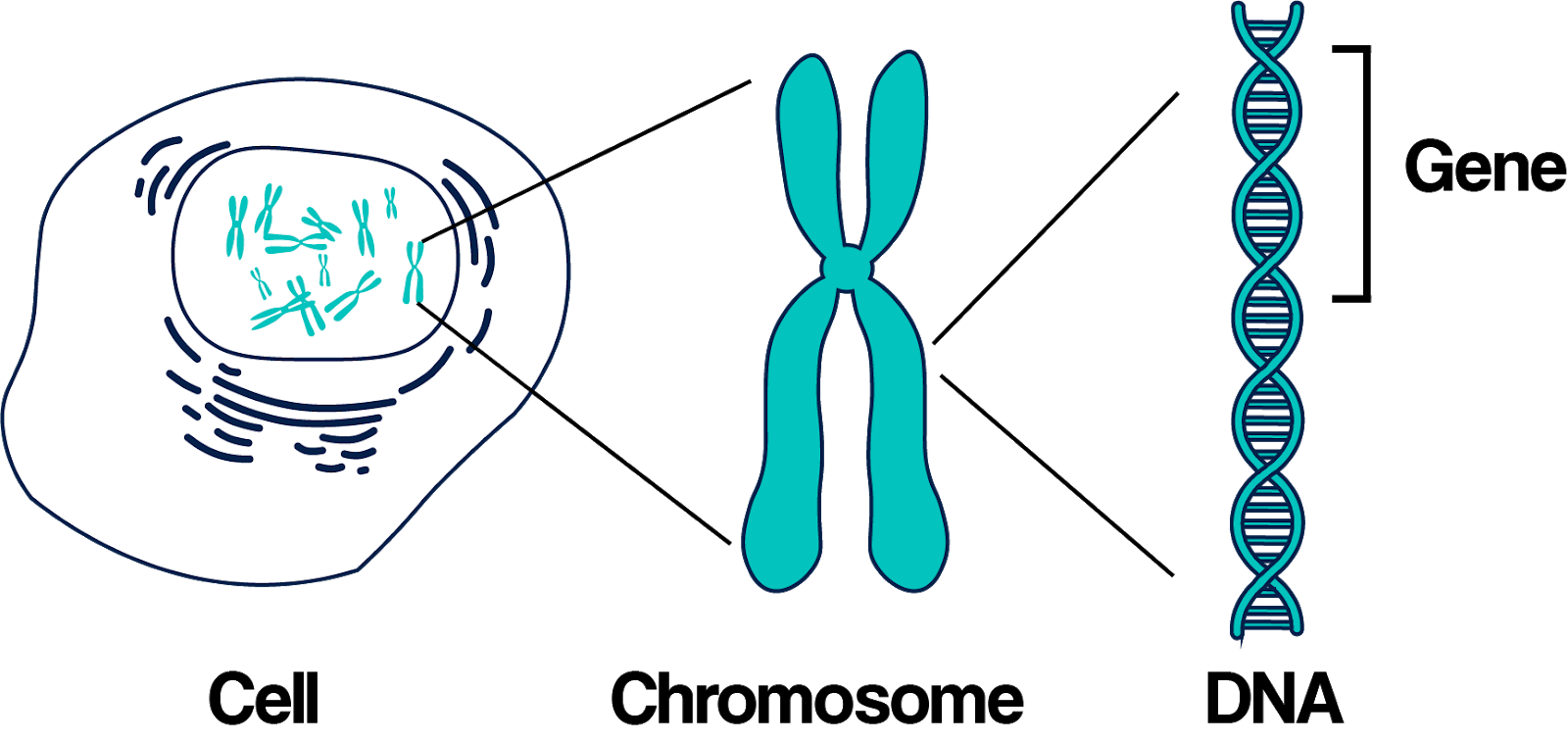
The genome consists of numerous fragments, which are called chromosomes.
Unfortunately it did not work. The chromosomes have different sizes, and therefore a different amount of information. This means that the tasks that Spark sent to the workers were not balanced and were performed slowly, because some nodes finished earlier and were idle. However, the tasks were completed. But when one SNP was requested, the imbalance again caused problems. The cost of processing SNP in larger chromosomes (that is, where we want to get data from) has decreased by only about 10 times. Many, but not enough.
And if you divide into even smaller partitions?
What I learned : never try to do 2.5 million partitions at all.
I decided to walk in full and partized every SNP. This ensured the same size of partitions. The bad was the idea . I used Glue and added the innocent line
partition_by = 'snp' . The task has started and started to run. A day later, I checked and saw that nothing was recorded in S3 so far, so I killed the task. It seems that Glue was writing intermediate files to a hidden place in S3, and many files, perhaps a couple of million. As a result, my mistake cost more than a thousand dollars and did not make my mentor happy.Partitioning + sorting
What I learned : sorting is still difficult, as well as setting up Spark.
The last attempt at partitioning was that I partitioned the chromosomes, and then sorted each partition. In theory, this would speed up each request, because the desired SNP data would have to be within several Parquet chunks within a given range. Alas, sorting even partitioned data has proven difficult. As a result, I switched to EMR for a custom cluster and used eight powerful instances (C5.4xl) and Sparklyr to create a more flexible workflow ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') ) ... but the task still has not been completed. I set it up differently: I increased the memory allocation for each query executor, I used nodes with a large amount of memory, I used broadcast variables (broadcasting variable), but every time it turned out to be half-measures, and gradually the performers began to fail, until everything stopped.
Becoming more creative
What I learned : sometimes special data requires special solutions.
Each SNP has a position value. This number corresponds to the number of bases lying along its chromosome. This is a good and natural way to organize our data. At first I wanted to partition into regions of each chromosome. For example, positions 1 - 2000, 2001 - 4000, etc. But the problem is that the SNPs are unevenly distributed across the chromosomes, so therefore the size of the groups will vary greatly.
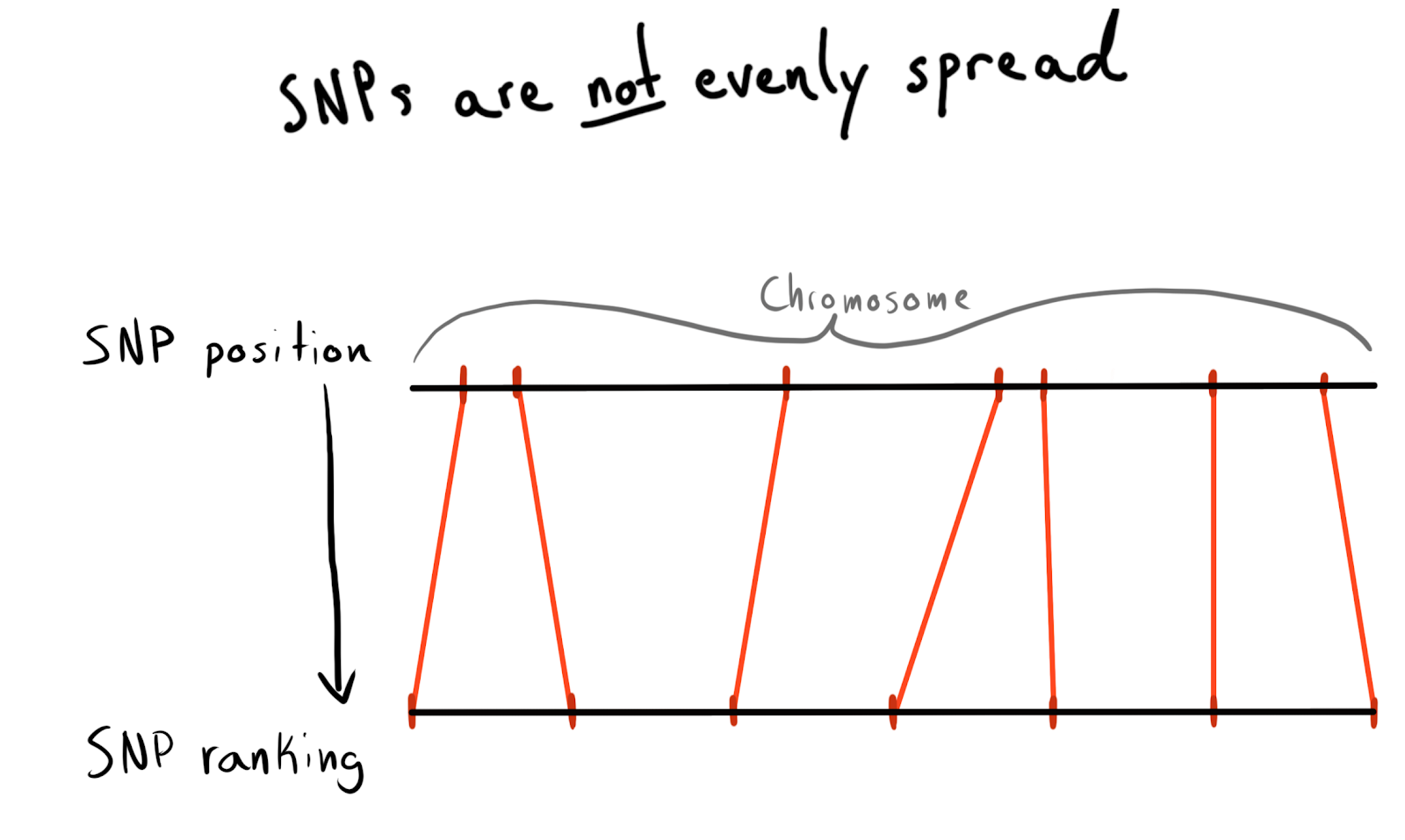
As a result, I came to a breakdown into categories (rank) positions. According to the data already loaded, I drove a request to get a list of unique SNPs, their positions and chromosomes. Then sorted the data inside each chromosome and collected the SNPs into groups (bin) of a given size. Let's say 1000 SNPs each. This gave me an SNP relationship with a chromosome-group.
In the end, I made groups (bin) for 75 SNPs, I will explain the reason below.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup() First attempt with Spark
What I learned : Spark integration is fast, but partitioning is still expensive.
I wanted to read this small (2.5 million lines) data frame in Spark, merge it with the raw data, and then partition it into the newly added
bin column. # Join the raw data with the snp bins data_w_bin <- raw_data %>% left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>% group_by(chr_bin) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr_bin') ) I used
sdf_broadcast() , so Spark finds out that it must send a data frame to all nodes. This is useful if the data is small and is required for all tasks. Otherwise, Spark tries to be smart and distributes data as needed, which can cause brakes.And again, my idea did not work: the tasks worked for some time, completed the merge, and then, like the executors started by partitioning, they began to fail.
Adding AWK
What I have learned : do not sleep when you are taught the basics. Surely someone already solved your problem back in the 1980s.
Up to this point, the cause of all my failures with Spark was the data jumble in the cluster. Perhaps the situation can be improved by preprocessing. I decided to try to divide the raw text data into chromosome columns, so I hoped to provide Spark with “pre-partitioned” data.
I looked at StackOverflow for how to break down the column values, and found such an excellent answer. With AWK, you can split a text file by column values by writing to the script, rather than sending the results to
stdout .For the sample, I wrote a Bash script. I downloaded one of the packed TSVs, then unpacked it with
gzip and sent it to awk . gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}' It worked!
Filling cores
What I learned :
gnu parallel is a magic thing, everyone should use it.The separation was rather slow, and when I ran
htop to test the use of a powerful (and expensive) EC2 instance, it turned out that I only use one core and about 200 MB of memory. To solve the problem and not lose a lot of money, you had to figure out how to parallelize the work. Fortunately, in the absolutely amazing Data Science book at the Command Line of Jeron Janssens, I found a chapter on parallelization. From it, I learned about gnu parallel , a very flexible method of implementing multithreading in Unix.
When I started the split using the new process, everything was fine, but there was a bottleneck - downloading S3 objects to disk was not too fast and not completely parallelized. To fix this, I did this:
- I found out that it is possible to realize the stage of S3-downloading directly in the pipeline, completely eliminating intermediate storage on the disk. This means that I can avoid writing raw data to disk and using even smaller, and therefore cheaper, storage on AWS.
aws configure set default.s3.max_concurrent_requests 50command,aws configure set default.s3.max_concurrent_requests 50greatly increased the number of threads that AWS CLI uses (10 by default).- I switched to a speed-optimized network instance EC2, with the letter n in the name. I found that the loss of computational power when using n-instances is more than offset by an increase in download speed. For most tasks, I used c5n.4xl.
- Changed
gziptopigz, this is a gzip tool that can do cool stuff for parallelization of the initially not parallelized task of unpacking files (this helped the least).
# Let S3 use as many threads as it wants aws configure set default.s3.max_concurrent_requests 50 for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do aws s3 cp s3://$batch_loc$chunk_file - | pigz -dc | parallel --block 100M --pipe \ "awk -F '\t' '{print \$1\",...\"$30\">\"chunked/{#}_chr\"\$15\".csv\"}'" # Combine all the parallel process chunks to single files ls chunked/ | cut -d '_' -f 2 | sort -u | parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}' # Clean up intermediate data rm chunked/* done These steps are combined with each other so that everything works very quickly. Due to the increase in download speed and the rejection of writing to disk, I could now process a 5-terabyte package in just a few hours.
This tweet should have mentioned 'TSV'. Alas.
Using re-parsed data
What I learned : Spark loves uncompressed data and does not like to combine partitions.
Now the data was in S3 in an unpacked (read, shared) and semi-ordered format, and I could return to Spark again. A surprise was waiting for me: I again failed to achieve the desired! It was very difficult to tell Spark exactly how the data is partized. And even when I did it, it turned out that there were too many partitions (95 thousand), and when I reduced the number to reasonable limits with the help of
coalesce , it ruined my partitioning. I am sure this can be fixed, but in a couple of days of searching I could not find a solution. In the end, I completed all the tasks in Spark, although it took some time, and my separated Parquet files were not very small (~ 200 Kb). However, the data lay where needed.
Too small and uneven, wonderful!
Testing local Spark requests
What I learned : Spark has too much overhead in solving simple problems.
Having downloaded the data in a thoughtful format, I was able to test the speed. I set up a script for R to start a local Spark server, and then I loaded the Spark data frame from the specified Parquet group repository (bin). I tried to load all the data, but could not get Sparklyr to recognize the partitioning.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time) The execution took 29.415 seconds. Much better, but not too good for mass testing anything. In addition, I could not speed up work using caching, because when I tried to cache a data frame in memory, Spark always fell, even when I allocated more than 50 GB of memory for a dataset that weighed less than 15.
Return to AWK
What I learned : associative arrays in AWK are very effective.
I realized that I can achieve a higher speed. I remembered that in the awesome AWK tutorial of Bruce Barnett I read about a cool feature called “ associative arrays ”. In fact, these are key-value pairs, which for some reason were named differently in AWK, and therefore I somehow did not even mention them. Roman Chepliak recalled that the term “associative arrays” is much older than the term “key-value pair”. Even if you search for a key value in Google Ngram , you will not see this term there, but you will find associative arrays! In addition, the "key-value pair" is most often associated with databases, so it is much more logical to compare with hashmap. I realized that I can use these associative arrays to associate my SNPs with a group table (bin table) and raw data without Spark.
For this, in the AWK script I used the
BEGIN block. This is a piece of code that is executed before the first data line is transferred to the main body of the script. join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" } The
while(getline...) command loaded all the lines from the CSV group (bin), set the first column (SNP name) as the key for the associative array bin and the second value (group) as the value. Then, in the { } block that is executed for all lines of the main file, each line is sent to the output file, which receives a unique name depending on its group (bin): ..._bin_"bin[$1]"_...The
batch_num and chunk_id corresponded to the data provided by the pipeline, which made it possible to avoid a race condition, and each execution thread running parallel wrote to its own unique file.Since I scattered all the raw data in folders on the chromosomes left over from my previous experiment with AWK, I could now write another Bash script to process on the chromosome at a time and give the S3 deeper partitioned data.
DESIRED_CHR='13' # Download chromosome data from s3 and split into bins aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv' | parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=\""$DESIRED_CHR"\" -v chunk=\"{}\" -f split_on_chr_bin.awk" # Combine all the parallel process chunks to single files and upload to rds using R ls chunked/ | cut -d '_' -f 4 | sort -u | parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds" rm chunked/* The script has two
parallel sections.In the first section, data is read from all files containing information on the desired chromosome, then this data is distributed into streams that spread the files into the corresponding groups (bin). To avoid a race condition, when several streams are written to a single file, AWK sends the file names for writing data to different places, for example,
chr_10_bin_52_batch_2_aa.csv . As a result, a lot of small files are created on the disk (for this I used terabyte EBS volumes).The pipeline from the second section of
parallel passes through the groups (bin) and combines their individual files into shared CSV with cat , and then sends them for export.Broadcasting in R?
What I learned : you can access
stdin and stdout from an R-script, and therefore use it in a pipeline.In the Bash script, you might notice this line:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... It translates all concatenated group files (bin) into the following R script. {} is a special parallel technique that any data it sends to the specified stream inserts data directly into the command itself. The {#} option provides a unique ID of the execution thread, and {%} is the slot number of the task (repeated, but never at the same time). A list of all options can be found in the documentation. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination ) When the variable
file("stdin") transferred to readr::read_csv , the data translated into the R script is loaded into the frame, which is then written to S3 directly in the form of a .rds file using aws.s3 .RDS is something like the younger version of Parquet, with no frills column storage.
After completing the Bash script, I received a bundle of
.rds files in S3, which allowed me to use efficient compression and built-in types.Despite the use of brake R, everything worked very quickly. Not surprisingly, the fragments on R that are responsible for reading and writing data are well optimized. After testing on one medium-size chromosome, the task was completed on the C5n.4xl instance in about two hours.
S3 restrictions
What I learned : thanks to the smart implementation of the paths, S3 can process many files.
I was worried if S3 would be able to process the many files transferred to it. I could make the file names meaningful, but how will S3 search for them?

S3 ,
/ . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate( # Extract chromosome from the file name chr = str_extract(Key, 'chr.{1,4}\\.csv') %>% str_remove_all('chr|\\.csv') ) %>% group_by(chr) %>% summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB # A tibble: 27 x 2 chr total_size <chr> <dbl> 1 0 163. 2 1 967. 3 10 541. 4 11 611. 5 12 542. 6 13 364. 7 14 375. 8 15 372. 9 16 434. 10 17 443. # … with 17 more rows , , ,
num_jobs , . num_jobs <- 7 # How big would each job be if perfectly split? job_size <- sum(chr_sizes$total_size)/7 shuffle_job <- function(i){ chr_sizes %>% sample_frac() %>% mutate( cum_size = cumsum(total_size), job_num = ceiling(cum_size/job_size) ) %>% group_by(job_num) %>% summarise( job_chrs = paste(chr, collapse = ','), total_job_size = sum(total_size) ) %>% mutate(sd = sd(total_job_size)) %>% nest(-sd) } shuffle_job(1) # A tibble: 1 x 2 sd data <dbl> <list> 1 153. <tibble [7 × 3]> purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1) , . Bash-
for . 10 . , . , . for DESIRED_CHR in "16" "9" "7" "21" "MT" do # Code for processing a single chromosome fi :
sudo shutdown -h now … ! AWS CLI
user_data Bash- . , . aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>> !
: API .
- . , . API .
.rds Parquet-, , . R-., ,
get_snp . pkgdown , .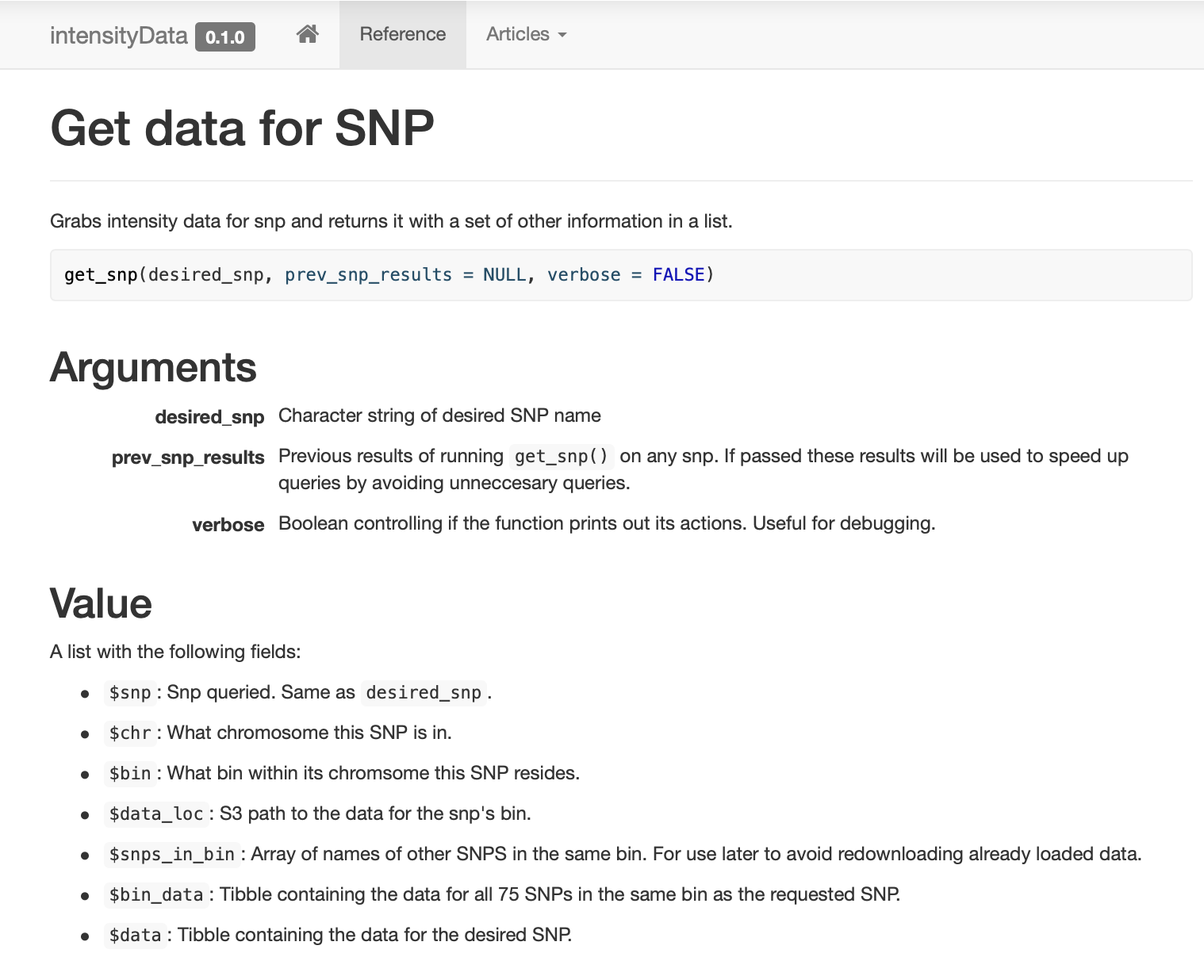
: , !
SNP , (binning) . SNP, (bin). ( ) .
# Part of get_snp() ... # Test if our current snp data has the desired snp. already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin if(!already_have_snp){ # Grab info on the bin of the desired snp snp_results <- get_snp_bin(desired_snp) # Download the snp's bin data snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc) } else { # The previous snp data contained the right bin so just use it snp_results <- prev_snp_results } ... , . , . ,
dplyr::filter , , .,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) : # Get bin-mates snps_in_bin <- my_snp_results$snps_in_bin for(current_snp in snps_in_bin){ my_snp_results <- get_snp(current_snp, my_snp_results) # Do something with results } results
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Conclusion
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel— , ;- Spark ;
- Spark ;
- AWK ;
stdinstdoutR-, ;- S3 ;
- — ;
- , ;
- API ;
- , !
Source: https://habr.com/ru/post/456392/
All Articles