Developing proteins in the cloud using Python and Transcriptic or How to create any protein for $ 360
What if you have an idea for a cool, healthy protein, and you want to get it in reality? For example, do you want to create a vaccine against H. pylori (like the Slovenian team at iGEM 2008 ) by creating a hybrid protein that combines E. coli flagellin fragments that stimulate the immune response with H. pylori flagellin?
 Design of hybrid flagellin for H. pylori vaccine presented by the Slovenian team at iGEM 2008
Design of hybrid flagellin for H. pylori vaccine presented by the Slovenian team at iGEM 2008
Surprisingly, we are very close to creating any protein we want, without leaving the Jupyter notebook, thanks to the latest developments in genomics, synthetic biology and, more recently, in cloud laboratories.
')
In this article, I will show the Python code from the idea of a protein to its expression in a bacterial cell, without touching the pipette or talking to any other person. The total cost will be only a few hundred dollars! Using the terminology of Vijay Pande of A16Z , this is Biology 2.0.
More specifically, in the article, the Python code of the cloud lab does the following:
First, the general Python settings that are needed for any Jupyter notepad. We import some useful Python modules and create some utility functions, mainly for data visualization.
Like AWS or any computing cloud, the cloud lab has molecular biology equipment, as well as robots that it leases over the Internet. You can issue instructions to your robots by clicking a few buttons in the interface or by writing code that programs them yourself. It is not necessary to write your own protocols, as I will do here, a significant part of molecular biology is standard routine tasks, so it is usually better to rely on a reliable alien protocol that has shown good interaction with robots.
Recently, a number of companies with cloud laboratories have appeared: Transcriptic , Autodesk Wet Lab Accelerator (beta, and built on the basis of Transcriptic), Arcturus BioCloud (beta), Emerald Cloud Lab (beta), Synthego (not yet launched). There are even companies built on top of cloud labs, such as Desktop Genetics , which specializes in CRISPR. Scientific articles about the use of cloud laboratories in real science are beginning to appear.
At the time of writing this article, only Transcriptic is in open access, so we will use it As I understand it, most of the Transcriptic business is built on automating common protocols, and writing your own protocols in Python (as I will do in this article) is less common.

Transcriptic "working cell" with refrigerators below and various laboratory equipment on the stand
I'll give Transcriptic robots instructions for the autoprotocol . Autoprotocol is a JSON-based language for writing protocols for laboratory robots (and people, as it were). Autoprotocol is mostly done on this Python library . The language was originally created and is still supported by Transcriptic, but, as I understand it, it is completely open. There is good documentation .
An interesting idea is that on the autoprotocol you can write instructions for people in remote laboratories — say, in China or India — and potentially get some advantages from using both people (their judgment) and robots (no judgment). We need to mention protocols.io here , this is an attempt to standardize protocols to increase reproducibility, but for humans, not robots.
Example autoprotocol snippet
In addition to importing standard libraries, I will need some specific molecular biological utilities. This code is mainly for autoprotocol and transcriptic.
In the code, the concept of "dead volume" is often found. This means the last drop of liquid that Transcriptic robots cannot take with a pipette from test tubes (because they don’t see!). You have to spend a lot of time to make sure that there is a sufficient amount of material in the flasks.
Despite the connection with modern synthetic biology, DNA synthesis is a rather old technology. For decades we have been able to make olikonucleotides (that is, DNA sequences up to 200 bases). However, it was always expensive, and chemistry never allowed for long DNA sequences. Recently it has become possible to synthesize whole genes (up to thousands of bases) at a reasonable price. This achievement really opens the era of "synthetic biology".
Craig Venter's Synthetic Genomics has advanced synthetic biology the furthest by synthesizing an entire body — over a million bases in length. As the length of the DNA increases, the problem becomes not synthesis, but assembly (i.e., stitching together the synthesized DNA sequences). With each assembly, you can double the length of DNA (or more), so after a dozen or so iterations a rather long molecule is obtained! The distinction between synthesis and assembly should soon become clear to the end user.
The price of DNA synthesis falls quite quickly, from more than $ 0.30 per basis two years ago to about $ 0.10 today, but it develops more like bacteria than processors. In contrast, DNA sequencing prices fall faster than Moore's law. A target of $ 0.02 per base is scheduled as an inflection point , where you can replace a lot of time consuming manipulations with DNA with simple synthesis. For example, at this price you can synthesize a whole plasmid of 3kb for $ 60 and skip a bunch of molecular biology. Hopefully we will achieve this in a couple of years.
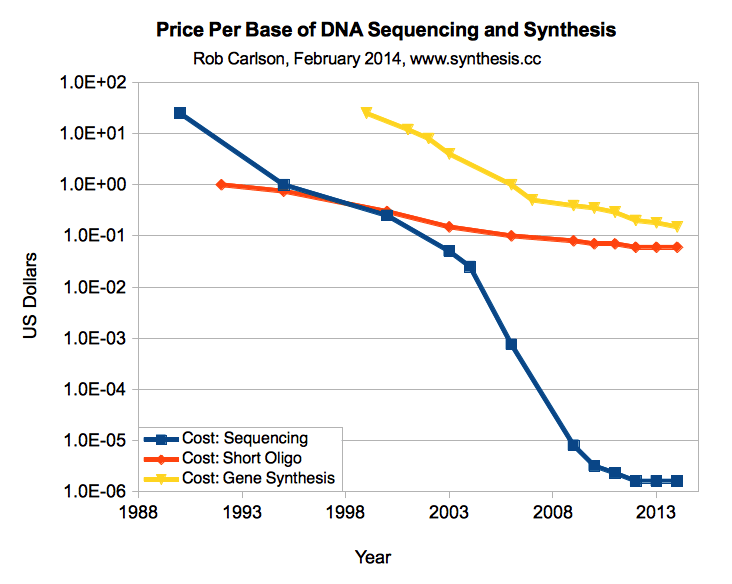
Prices for DNA synthesis compared to prices for DNA sequencing, price for 1 basis (Carlson, 2014)
There are several large companies in the field of DNA synthesis: IDT is the largest producer of oliconucleotides, and can also produce longer (up to 2kb) “gene fragments” ( gBlocks ). Gen9 , Twist, and DNA 2.0 usually specialize in longer DNA sequences — these are gene synthesis companies. There are also some interesting new companies, such as Cambrian Genomics and Genesis DNA , which are working on the next generation synthesis methods.
Other companies, such as Amyris , Zymergen and Ginkgo Bioworks , use the DNA synthesized by these companies to work at the organism level. Synthetic Genomics also does this, but it itself synthesizes DNA.
Ginkgo recently made a deal with Twist to make 100 million bases: the biggest deal I saw. This proves that we live in the future, Twist even advertised the promotional code on Twitter: when you buy 10 million bases of DNA (almost the entire yeast genome!), You get another 10 million for free.

Twist Niche Suggestion on Twitter
In this experiment, we synthesize a DNA sequence for a simple, green fluorescent protein (GFP). GFP protein was first found in a jellyfish that fluoresces under ultraviolet light. It is an extremely useful protein because it is easy to detect its expression by simply measuring the fluorescence. There are variations of GFP that produce yellow, red, orange, and other colors.
It is interesting to see how different mutations affect protein color, and this is a potentially interesting machine learning problem. Most recently, this would have to spend a lot of time in the lab, but now I will show you that it is (almost) as easy as editing a text file!
Technically, my GFP is a superfolder variant (sfGFP) with some mutations to improve qualities.
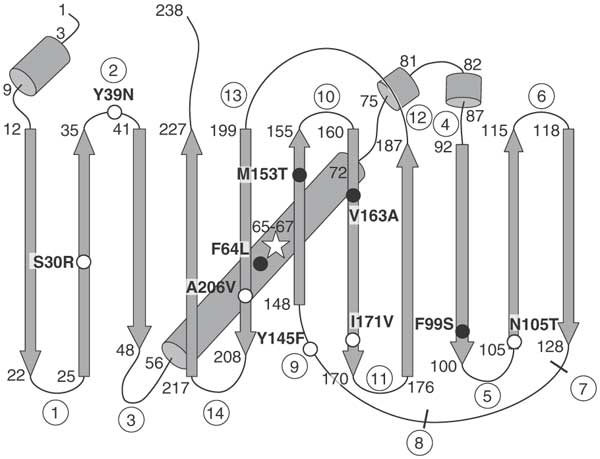
In superfolder-GFP (sfGFP), some mutations give it certain beneficial properties.
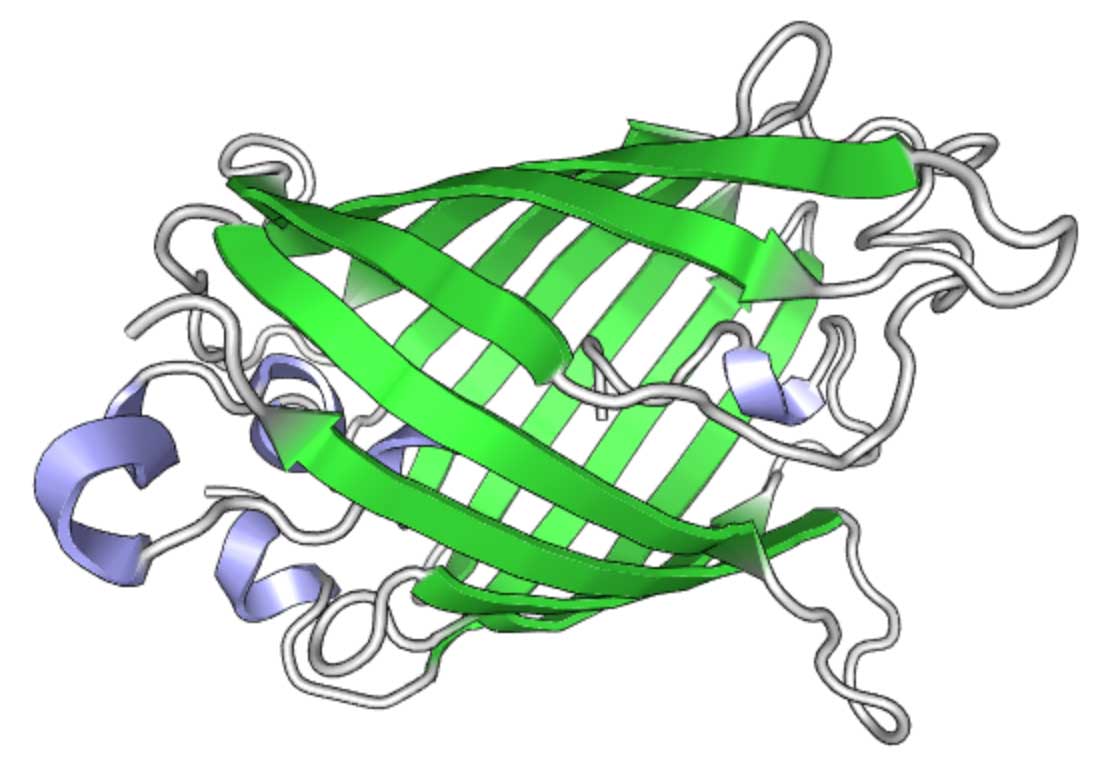
GFP structure (rendered using PV )
I was lucky to get into the Twist alpha testing program, so I used their DNA synthesis service (they kindly placed my tiny order — thanks, Twist!). This is a new company in our field, with a new simplified synthesis process. Prices are around $ 0.10 for the base or lower , but they are still in beta , and the alpha program in which I participated has closed. Twist raised about $ 150 million, so their technology is lively enthusiastic.
I sent my DNA sequence to Twist as an Excel spreadsheet (there is no API yet, but I guess it will be soon), and they sent the synthesized DNA directly to my box at the Transcriptic lab (I also used IDT for synthesis, but they didn't send DNA right in Transcriptic, which spoils the fun a bit).
Obviously, this process has not yet become a typical use case and requires some support, but it worked, so the entire pipeline remains virtual. Without this, I probably would need access to the laboratory — many companies would not send DNA or reagents to their home address.
GFP is harmless, so any kind of light is highlighted
To express this protein in bacteria, the gene needs to live somewhere, otherwise the synthetic DNA encoding the gene simply instantly degrades. As a rule, in molecular biology we use a plasmid, a piece of circular DNA that lives outside the bacterial genome and expresses proteins. Plasmids are a convenient way for bacteria to share useful, autonomous functional modules, such as antibiotic resistance. There can be hundreds of plasmids in a cell.
A widely used terminology is that the plasmid is a vector , and synthetic DNA is an insertion. So here we are trying to clone the insertion into a vector, and then transform the bacteria with a vector.
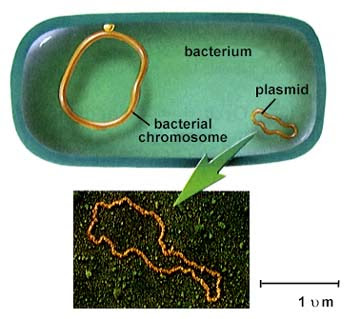
Bacterial genome and plasmid (not to scale!) ( Wikipedia )
I chose a fairly standard plasmid in the pUC19 series. This plasmid is very often used, and since it is available as part of the standard Transcriptic inventory, we do not need to send anything to them.
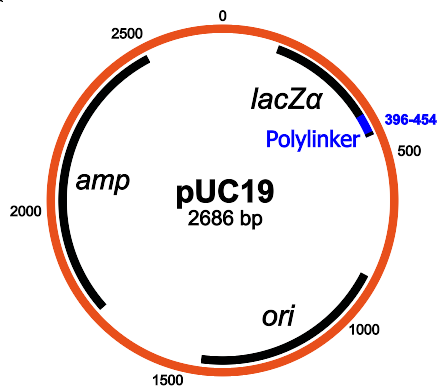
Structure of pUC19: the main components are the ampicillin resistance gene, lacZα, MCS / polylinker and the origin of replication (Wikipedia)
PUC19 has a pleasant function: since it contains the lacZα gene, you can use the blue-white selection method on it and see which colonies successfully inserted. Two chemicals are needed: IPTG and X-gal , and the scheme works as follows:
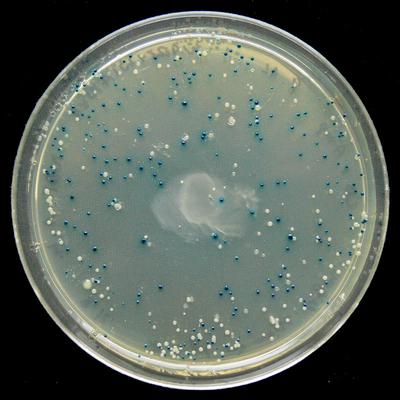
Blue-white selection shows where lacZα expression has been deactivated ( Wikipedia )
The openwetware documentation says:
It is easy to obtain a DNA sequence for sfGFP by taking a protein sequence and encoding it with codons suitable for the host organism (here, E. coli ). This is a medium-sized protein with 236 amino acids, so at 10 cents per base, DNA synthesis costs about $ 70 .
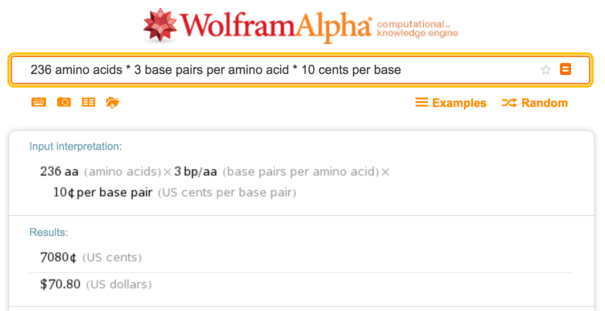
Wolfram Alpha, the calculation of the cost of synthesis
The first 12 bases of our sfGFP is the Shine – Dalgarno sequence , which I added myself, which theoretically should increase the expression (AGGAGGACAGCT, then ATG ( start codon ) starts the protein). According to the computational tool developed by Salis Lab ( lecture slides ), we can expect medium to high expression of our protein (translation initiation rate is 10,000 "arbitrary units").
First, I check that the sequence pUC19, which I downloaded from the NEB , is the correct length and includes the expected polylinker .
We make some basic QC to make sure that EcoRI and BamHI are present in pUC19 only once (the following restriction enzymes are present in the Transcriptic inventory by default: PstI , PvuII , EcoRI , BamHI , BbsI , BsmBI ).
Now we look at the lacZα sequence and check that there is nothing unexpected. For example, it must begin with Met and end with a stop codon. It is also easy to confirm that this is a full 324bp lacZα ORF by downloading the sequence pUC19 to the free snapgene viewer .
DNA assembly simply means stitching the fragments. Usually you collect several DNA fragments into a longer segment, and then clone it into a plasmid or genome. In this experiment, I want to clone one DNA segment into plasmid pUC19 downstream of the lac promoter for expression in E. coli .
There are many ways to clone (for example, NEB , openwetware , addgene ). Here I will use the Gibson assembly ( developed by Daniel Gibson in Synthetic Genomics in 2009), which is not necessarily the cheapest method, but simple and flexible. You only need to place the DNA that you want to collect (with appropriate overlaps) in a tube with the Gibson Assembly Master Mix, and it is going to be on its own!
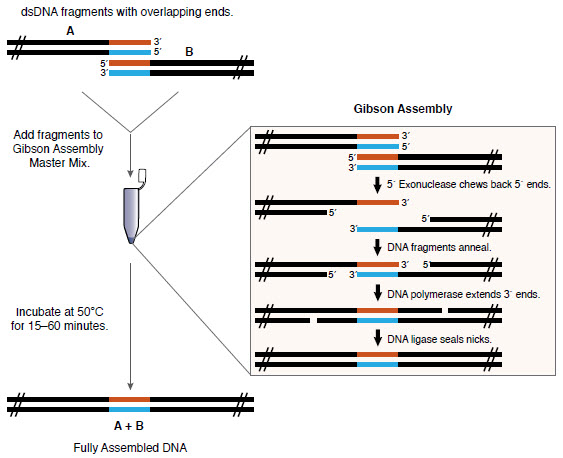
Gibson Assembly Review ( NEB )
We start with 100 ng of synthetic DNA in 10 μl of liquid. This equals 0.21 picomoles of DNA or a concentration of 10 ng / μl.
According to the NEB assembly protocol , this is enough source material:
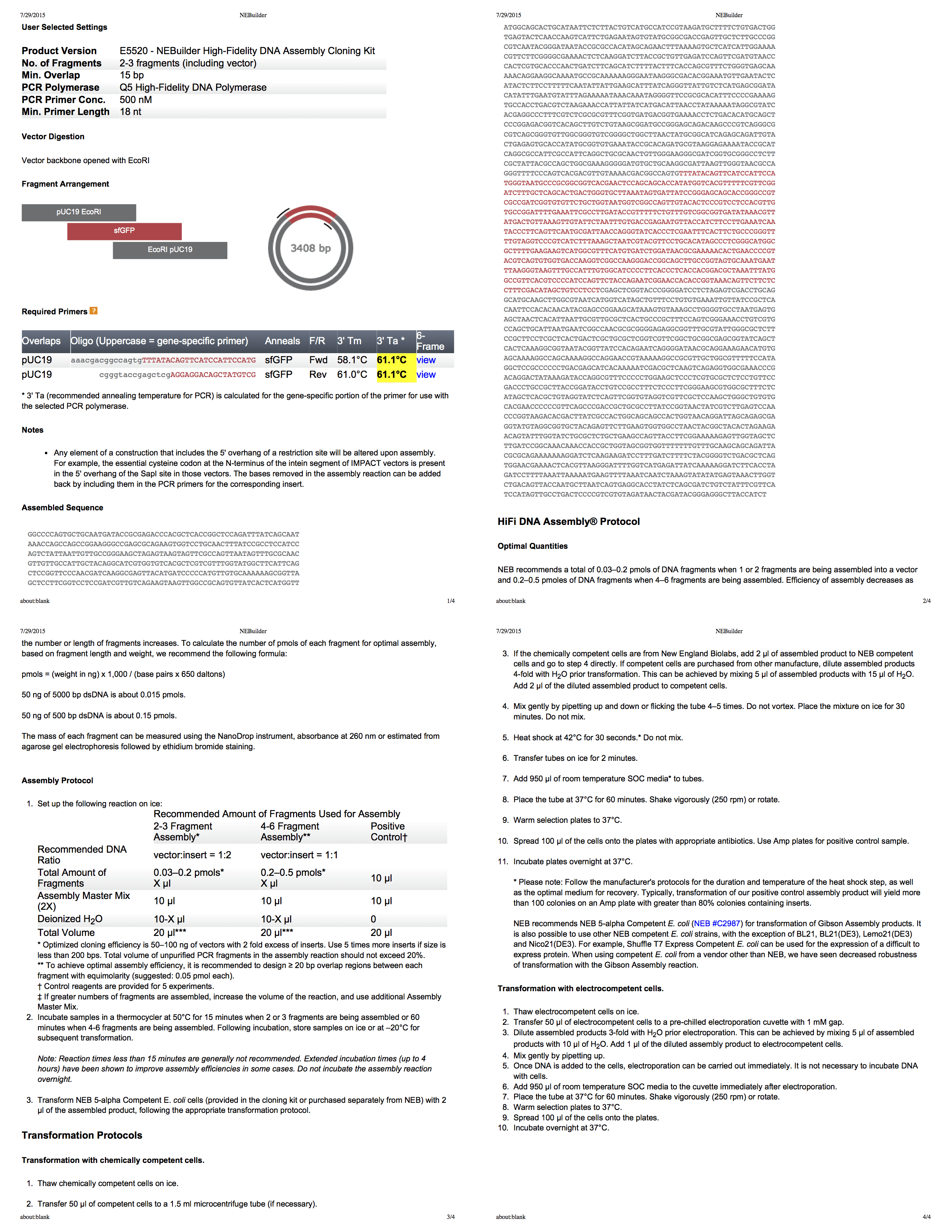
Biolab's NEBuilder is a really great tool for creating a Gibson assembly protocol. PDF . pUC19 EcoRI, PCR [, — . .] .
:
The Gibson assembly depends on the DNA sequence that you are assembling, having some overlapping sequence (see the NEB protocol above for detailed instructions). In addition to simple amplification, PCR also allows you to add a flanking DNA sequence by simply inserting an additional sequence into the primers (you can also clone using only OE-PCR ).
We synthesize primers according to the NEB protocol above. I tried the Quickstart protocol on the Transcriptic website, but there is also an auto protocol command . Transcriptic , 1-2 ( , - , ).
IDT OligoAnalyzer . PCR primer dimer , NEB .
I went through many PCR iterations before getting satisfactory results, including experiments with several different brands of PCR mixes. Since each of these iterations can take several days (depending on the length of the queue to the laboratory), it is worthwhile to spend time in advance for debugging: it saves a lot of time in the long run. As the capacity of the cloud lab increases, this problem should become less acute. However, your first protocol is unlikely to succeed - there are too many variables.
In the gel, you can estimate the correct size of the product after increasing the concentration (position of the strip in the gel) and the correct amount (darkness of the strip). The gel has a ladder corresponding to different lengths and quantities of DNA that can be used for comparison.
In the gel photograph below the band D1, E1, F1 contain, respectively, 2 μl, 4 μl and 8 μl of the amplified product. I can estimate the amount of DNA in each lane compared to DNA in the ladder (50 ng of DNA per lane in the ladder). I think the results look very clean.
I tried to use GelEval for image analysis and concentration assessment. , , , , . . GelEval 40 /.
, , dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
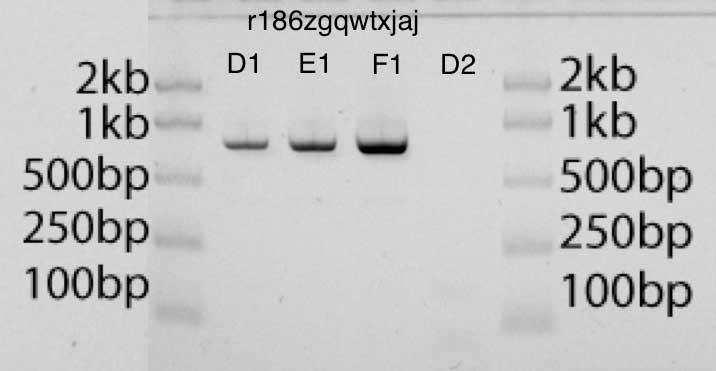
- EcoRI- pUC19, (D1, E1, F1), (D2)
Recently, Transcriptic began to provide interesting and useful diagnostic data from its robots. At the time of this writing, they are not available for download, so for now I only have an image of the temperatures during thermal cycling.
The data looks good, without unexpected peaks or valleys. A total of 35 PCR cycles, but some of these cycles are performed at a very high temperature within the PCR touchdown . In my previous attempts to amplify this segment - of which there were several! - there were problems with the hybridization of primers, so here PCR works a lot of time at high temperatures, which should improve the accuracy.

Thermocyclic diagnostics for a touchdown PCR: temperature block, sample and cover for 35 cycles and 42 minutes
To insert our sfGFP DNA into pUC19, you first need to cut the plasmid. Following the NEB protocol, I do this using the EcoRI restriction enzyme . In the standard Transcriptic inventory there are reagents that I need: these are the NEB EcoRI and 10x CutSmart buffer , as well as the NEB pUC19 plasmid .
For information, lower prices from their inventory. In fact, I pay only part of the price, since Transcriptic takes payment for the quantity actually consumed:
I followed the NEB protocol as much as possible:
I conducted this experiment twice in slightly different conditions and with gels of different sizes, but the results are almost identical. I like both gels.
Initially, I did not allocate enough space for a “dead” volume (in test tubes 1.5 ml there is a dead volume of 15 μl!). I think this explains the difference between D1 and E1 (these two bands should be identical). The dead volume problem is easily solved by creating a proper working stock of diluted EcoRI at the beginning of the protocol.
Despite this error, in both gels the bands D1 and E1 are strong bands in the correct position 2,6kb. On the D2 band, uncut plasmid: as expected, it is not visible in one gel and is barely visible in another.
Two gel photos look quite different. This is partly due to the fact that Transcriptic has not yet automated this step.
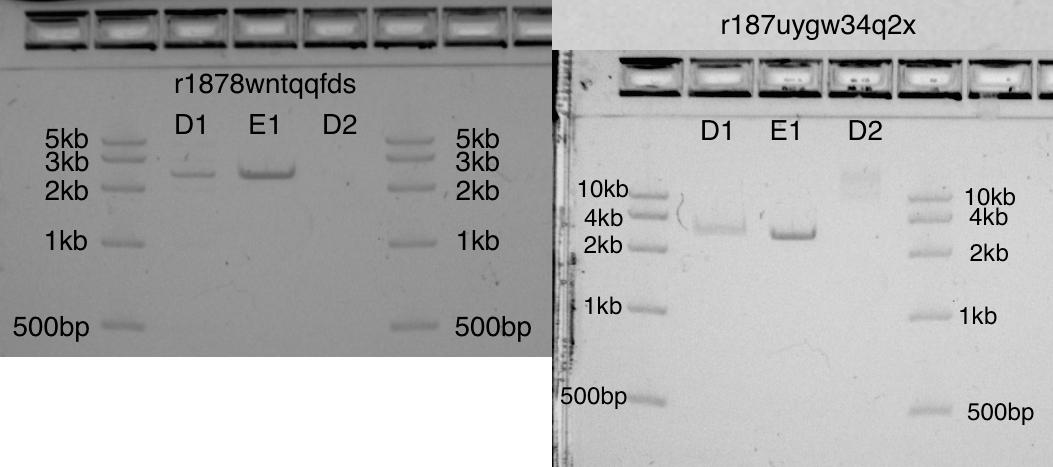
Two gels showing cut pUC19 (2,6kb) on the D1 and E1 bands, and uncut pUC19 on D2
, — , M13 ( ) qPCR , , . , , , .
, M13 , M13.
I can access qPCR data in JSON format via the Transcriptic API. This feature is not well documented , but can be extremely useful. The APIs even give you access to some diagnostic data from robots, which can aid in debugging.
First, we request the launch data:
Then we specify this id to get the qPCR “post-processing” data:
Here are the Ct values (cycle threshold) for each tube. Ct is just the point at which fluorescence exceeds a certain value. She roughly says how much DNA there is at the moment (and, therefore, approximately where we started).
, D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
, qPCR , . v3 , v1, .
The gel is also very clean, shows strong bands just below 1kb in bands B2, B4, B6, D2, D4, D6: this is exactly the size we expect (insertion is about 740bp, and primers M13 are about 40bp up and down). The second bar corresponds to the primers. You can be sure of this, since the F2 and F4 bands contain only primer DNA.
Polyacrylamide gel electrophoresis: with the Gibson v3 assembly shows stronger bands (D2, D4, D6), in accordance with the qPCR data given above
Transformation is the process of changing the body by adding DNA. In this experiment, we transform E. coli using the sfGFP-expressing plasmid pUC19.
We use an easy-to-work strain Zymo DH5α Mix & Go and the recommended protocol . This strain is part of the standard Transcriptic inventory. In general, transformations can be complex, since competent cells are quite fragile, so the simpler and more reliable the protocol, the better. In ordinary molecular biology laboratories, these competent cells would probably be too expensive for general use.
Zymo Mix & Go cells with simple protocol
This protocol is a good example of how difficult it is to adapt human protocols for use by robots and how it may unexpectedly fail. Protocols are sometimes surprisingly vague (“shake the tube from side to side”), based on the general context of molecular biologists, or they can suddenly request advanced image processing (“make sure the sediment is stirred”). People do not mind such tasks, but the robots need clearer instructions.
. , 37°C. , , , , Transcriptic — , . , , - , . . .
There are usually reasonable solutions: sometimes you just need to use different reagents (for example, more durable cells, such as Mix & Go above); sometimes you just lay actions with a margin (for example, shake ten times instead of three); Sometimes you need to come up with special tricks for robots (for example, use a PCR machine for heat stroke).
Of course, the big advantage is that, once the protocol has worked once, you can generally rely on it again and again. You can even quantify how reliable the protocol is and improve it over time!
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
In the following photos, we see that without the antibiotic (the plate on the left), growth is observed on all six plates, although strongly to varying degrees, which causes concern. It seems that Transcriptic robots do not really cope with the uniform distribution, which requires some dexterity.
( ) , . , , , , 55 10 . . , . , , .
( , , , E. coli . Growth is much weaker on plates with ampicillin, although there are much more bacteria, as expected).
In general, the transformation worked well enough to continue, although there are some flaws.

Plates of cells transformed with pUC19, after 18 hours: without antibiotic (left) and with antibiotic (right)
Since the Gibson assembly and simple transformation of pUC19 seem to work, you can now try the transformation with a fully assembled plasmid expressing sfGFP.
In addition to the assembled insertion, I will also add some IPTG and X-gal to the plates to see a successful transformation using the blue-white selection method . This additional information is useful, because if a transformation with the usual pUC19, which does not contain sfGFP, goes through, it will still give resistance to antibiotics.
According to this table , sfGFP shines best at excitation wavelengths of 485 nm / 510 nm. I found that 485/535 works better in Transcriptic. I guess because 485 and 510 are too similar. I measure bacterial growth at 600 nm ( OD600 ).
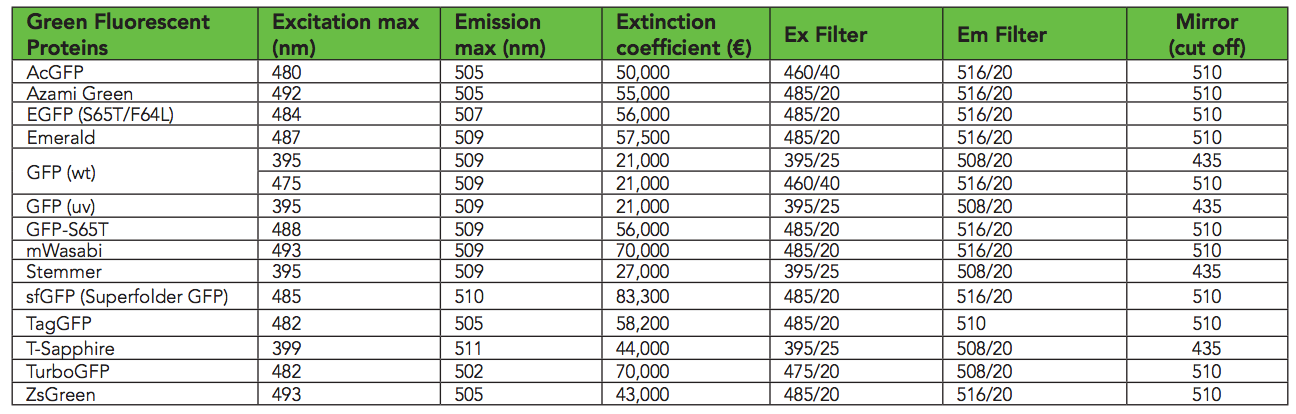
A variety of GFP ( biotek )
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
When colonies grow on an ampicillin plate, I can “collect” individual colonies and plant them on a 96-tube plate. For this, there is a special command ( autopick ) in the autoprotocol .
The blue-white screen perfectly showed mainly white colonies on plates with antibiotics (1-4) and only blue ones on plates without antibiotics (5-6). This is exactly what I expected, and I was glad to see it, especially since I used my own IPTG and X-gal, which I sent to Transcriptic.
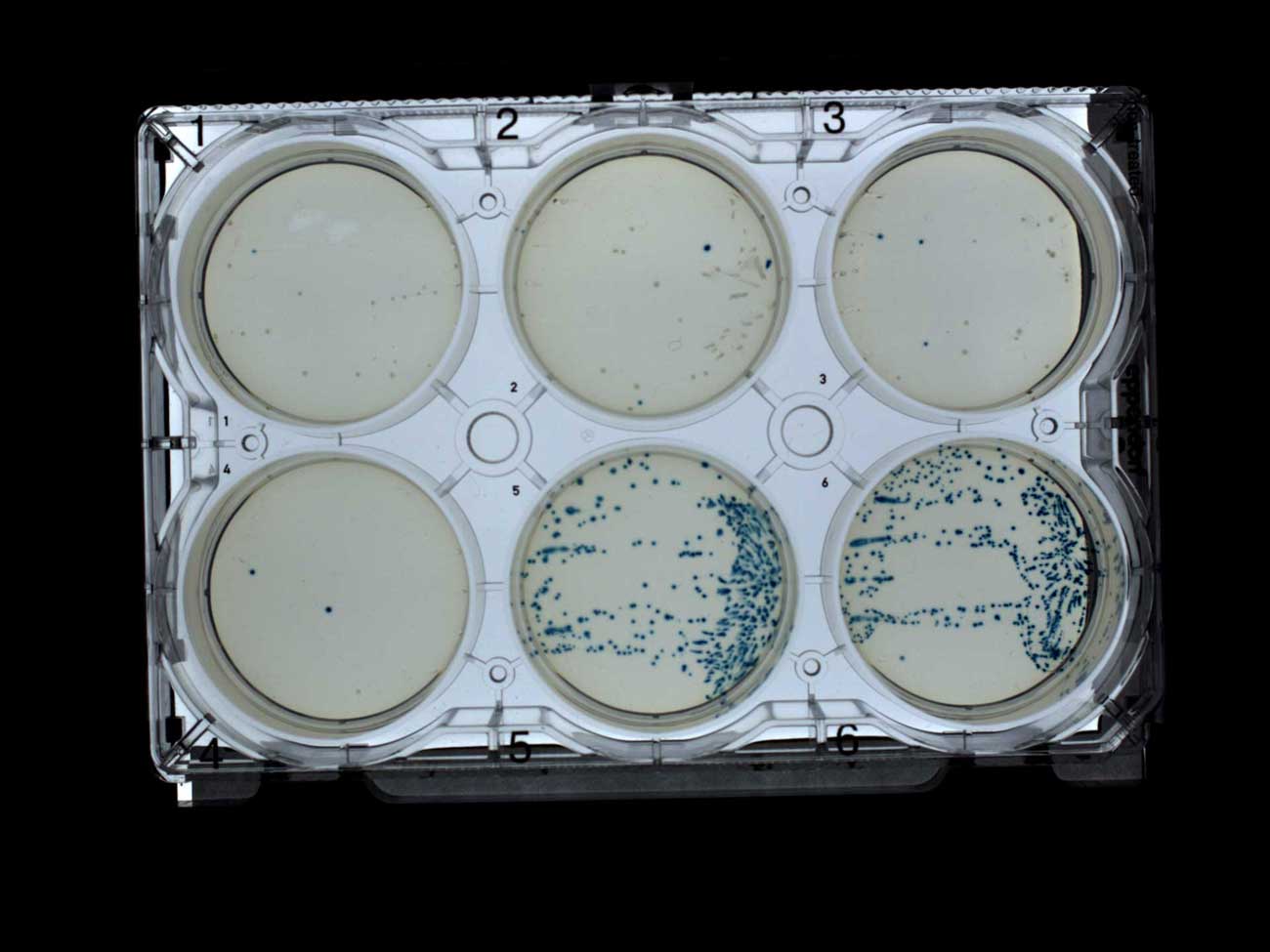
Screening using white-blue plate selection with ampicillin (1-4) and without antibiotic (5-6)
However, the robot-collector of colonies did not work well with these white-blue colonies. The image below is created by subtracting consecutive photographs of the plates after each round of plate selection and increasing the contrast of the differences (in GraphicsMagick ). In this way, I can visualize which colonies were collected (although not perfect, because the collected colonies are not completely removed).
I also signed the image with the number of colonies collected by the Transcriptic robot. It was assumed that he will collect a maximum of 10 colonies from the first five plates. However, several colonies were generally collected, and these are usually blue colonies. The robot only managed to find ten colonies on a control plate with only blue colonies. My working theory is that a robot collecting colonies preferably collects blue colonies, since they are more contrasting.

Screening plates for white-blue breeding with ampicillin (1-4) and without antibiotic (5-6), indicating the number of collected colonies
The blue-white screening served a specific purpose. He showed that most colonies are transformed correctly. At least there is an insertion. However, for a better collection of colonies, I repeated the experiment without X-gal.
Only with white colonies, the robot assembler successfully collected ten colonies from each of the first five plates. It can be assumed that in most of the collected colonies there are successful insertions.

Colonies growing on plates with ampicillin (1-4) and without antibiotic (5-6)
After growing 50 selected colonies on a 96-well plate for 20 hours, I measure the fluorescence to check for sfGFP expression. Transcriptic uses a Tecan Infinite reader to measure fluorescence and absorption (and luminescence, if you want) .
In theory, an assembled plasmid should be collected in any colony with growth, since it needs antibiotic resistance for growth, and each assembled plasmid expresses sfGFP. In fact, there are many reasons why this may not be the case, not least because you can lose the sfGFP gene from a plasmid without losing resistance to ampicillin. A bacterium that loses the sfGFP gene has an advantage in selection over its competitors, because it does not waste excess energy, and with a sufficient number of generations of growth, this will definitely happen.
I collect absorption (OD600) and fluorescence data every four hours for 20 hours (about 60 generations).
We place on the chart the data of the 20th hour and the traces of previous measurements. Actually, I am only interested in the latest data, since that is when the peak of fluorescence should be observed.
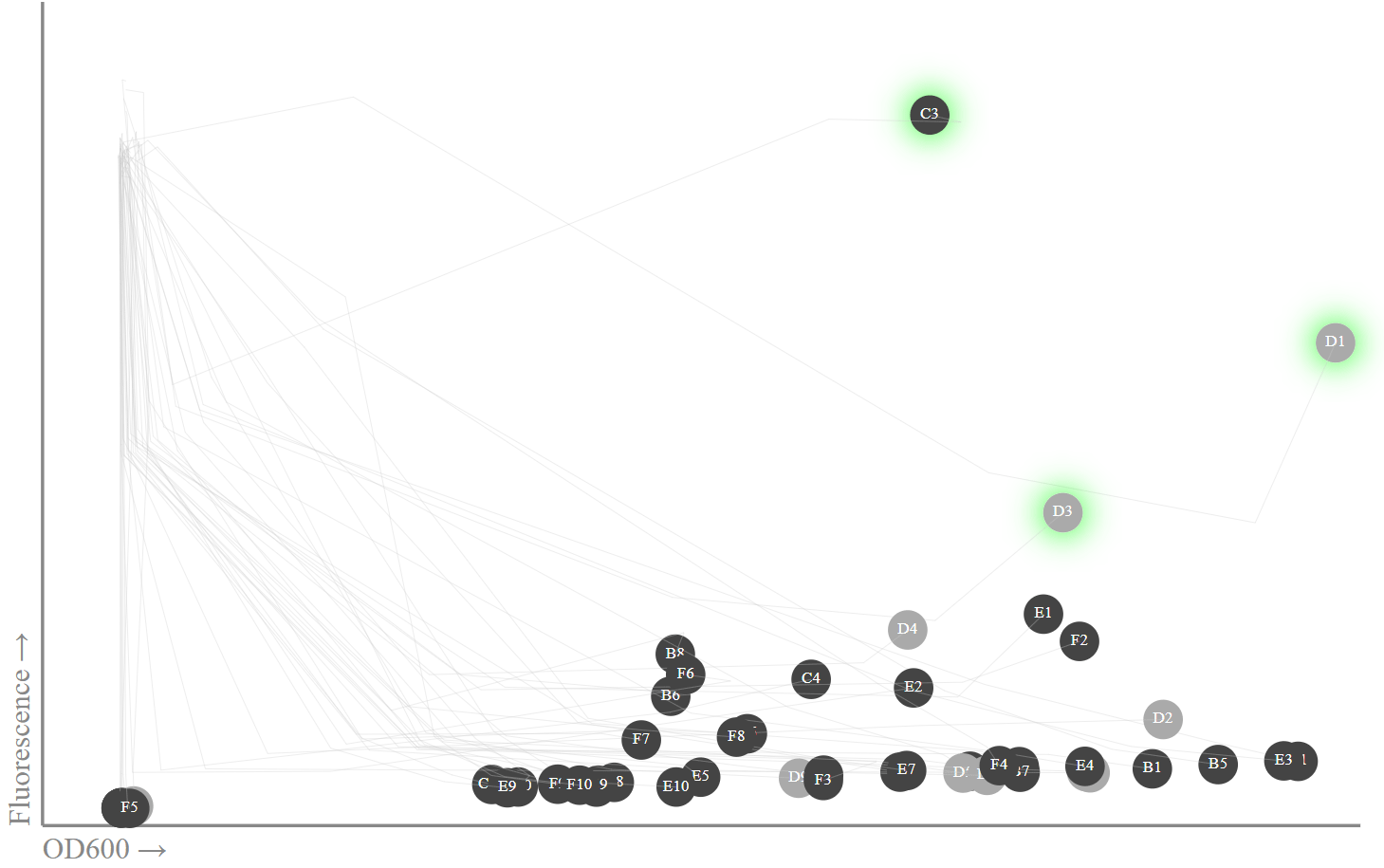
OD600: , . , sfGFP
miniprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
Based on fluorescence data and sequencing results, it appears that only three of the 50 colonies produce sfGFP and fluoresce. This is not as much as I expected. However, since three separate growth stages have passed (on a plate, in a test tube, for miniprep), about 200 generations of growth have undergone to this stage, so there were quite a few possibilities for mutations to occur.
There must be ways to make the process more efficient, especially since I am far from being an expert on these protocols. However, we successfully produced transformed cells with the expression of engineered GFP using only Python code!
Depending on how to measure, the cost of this experiment was about $ 360, not counting money for debugging:
I think that the cost can be reduced to $ 250-300 with some modifications. For example, a robotic collection of 50 colonies is suspiciously expensive and can probably be abandoned.
In my experience, this price seems high for some (molecular biologists) and low for others (people from IT). Since Transcriptic basically simply charges for reagents on the price list, the main cost difference is labor. The robot already costs quite cheaply per hour, and he is not averse to getting up in the middle of the night to photograph the plate. Once the protocols have been approved, it’s hard to imagine that even a graduate student will be cheaper, especially considering the opportunity costs.
, . , - , . , : , , IDT .
, . , :
Of course, there are some drawbacks, especially since the tools have just begun to develop. If we compare with the Internet, then we are in the area of 1994:
Although there is quite a lot of code and quite a lot of debugging, I think it is possible to create some kind of software that accepts a sequence of proteins at the input, and creates bacteria at the output with the expression of this protein.
For this to work, several things must happen:
In many applications, you also want to purify your protein (through a column ) or, perhaps, simply force bacteria to secrete it. Suppose that soon we can do this in a cloud lab, or that we can conduct in vivo experiments (i.e., inside a bacterial cell).
There are many possibilities for the protocol to actually work better than a human, for example: the design of promoters and RBS to optimize the expression specific to your sequence; experiment success probability statistics based on comparable experiments; automated gel analysis.
After all this, it may not be entirely clear why create such a protein. Here are some ideas:
For new ideas on what's possible when designing proteins, look at hundreds of iGEM projects .
In the end, I would like to thank Ben Miles of Transcriptic for helping to complete this project.
Surprisingly, we are very close to creating any protein we want, without leaving the Jupyter notebook, thanks to the latest developments in genomics, synthetic biology and, more recently, in cloud laboratories.
')
In this article, I will show the Python code from the idea of a protein to its expression in a bacterial cell, without touching the pipette or talking to any other person. The total cost will be only a few hundred dollars! Using the terminology of Vijay Pande of A16Z , this is Biology 2.0.
More specifically, in the article, the Python code of the cloud lab does the following:
- Synthesis of a DNA sequence that encodes any protein that I want.
- Cloning this synthetic DNA into a vector that can express it.
- Transformation of the bacteria with this vector and confirmation that expression occurs.
Python setup
First, the general Python settings that are needed for any Jupyter notepad. We import some useful Python modules and create some utility functions, mainly for data visualization.
Code
import re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"): # to match Image syntax if type(width)==type(1): width = "{}px".format(width) html = ["<table style='width:{}'><tr>".format(width)] if header is not None: html += ["<th>{}</th>".format(h) for h in header] + ["</tr><tr>"] for image in images: html.append("<td><img src='{}' /></td>".format(image)) html.append("</tr></table>") show_html(''.join(html)) def new_section(title, color="#66aa33", padding="120px"): style = "text-align:center;background:{};padding:{} 10px {} 10px;".format(color,padding,padding) style += "color:#ffffff;font-size:2.55em;line-height:1.2em;" return HTML('<div style="{}">{}</div>'.format(style, title)) # Show or hide text HTML(""" <style> .section { display:flex;align-items:center;justify-content:center;width:100%; height:400px; background:#6a3;color:#eee;font-size:275%; } .showhide_label { display:block; cursor:pointer; } .showhide { position: absolute; left: -999em; } .showhide + div { display: none; } .showhide:checked + div { display: block; } .shown_or_hidden { font-size:85%; } </style> """) # Plotting style plt.rc("axes", titlesize=20, labelsize=15, linewidth=.25, edgecolor='#444444') sns.set_context("notebook", font_scale=1.2, rc={}) %matplotlib inline %config InlineBackend.figure_format = 'retina' # or 'svg' Cloud labs
Like AWS or any computing cloud, the cloud lab has molecular biology equipment, as well as robots that it leases over the Internet. You can issue instructions to your robots by clicking a few buttons in the interface or by writing code that programs them yourself. It is not necessary to write your own protocols, as I will do here, a significant part of molecular biology is standard routine tasks, so it is usually better to rely on a reliable alien protocol that has shown good interaction with robots.
Recently, a number of companies with cloud laboratories have appeared: Transcriptic , Autodesk Wet Lab Accelerator (beta, and built on the basis of Transcriptic), Arcturus BioCloud (beta), Emerald Cloud Lab (beta), Synthego (not yet launched). There are even companies built on top of cloud labs, such as Desktop Genetics , which specializes in CRISPR. Scientific articles about the use of cloud laboratories in real science are beginning to appear.
At the time of writing this article, only Transcriptic is in open access, so we will use it As I understand it, most of the Transcriptic business is built on automating common protocols, and writing your own protocols in Python (as I will do in this article) is less common.
Transcriptic "working cell" with refrigerators below and various laboratory equipment on the stand
I'll give Transcriptic robots instructions for the autoprotocol . Autoprotocol is a JSON-based language for writing protocols for laboratory robots (and people, as it were). Autoprotocol is mostly done on this Python library . The language was originally created and is still supported by Transcriptic, but, as I understand it, it is completely open. There is good documentation .
An interesting idea is that on the autoprotocol you can write instructions for people in remote laboratories — say, in China or India — and potentially get some advantages from using both people (their judgment) and robots (no judgment). We need to mention protocols.io here , this is an attempt to standardize protocols to increase reproducibility, but for humans, not robots.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ] Example autoprotocol snippet
Python settings for molecular biology
In addition to importing standard libraries, I will need some specific molecular biological utilities. This code is mainly for autoprotocol and transcriptic.
In the code, the concept of "dead volume" is often found. This means the last drop of liquid that Transcriptic robots cannot take with a pipette from test tubes (because they don’t see!). You have to spend a lot of time to make sure that there is a sufficient amount of material in the flasks.
Code
import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref # "Link a ref name (string) to a Container instance." import requests import logging # Transcriptic authorization org_name = 'hgbrian' tsc_headers = {k:v for k,v in json.load(open("auth.json")).items() if k in ["X_User_Email","X_User_Token"]} # Transcriptic-specific dead volumes _dead_volume = [("96-pcr",3), ("96-flat",25), ("96-flat-uv",25), ("96-deep",15), ("384-pcr",2), ("384-flat",5), ("384-echo",15), ("micro-1.5",15), ("micro-2.0",15)] dead_volume = {k:Unit(v,"microliter") for k,v in _dead_volume} def init_inventory_well(well, headers=tsc_headers, org_name=org_name): """Initialize well (set volume etc) for Transcriptic""" def _container_url(container_id): return 'https://secure.transcriptic.com/{}/samples/{}.json'.format(org_name, container_id) response = requests.get(_container_url(well.container.id), headers=headers) response.raise_for_status() container = response.json() well_data = container['aliquots'][well.index] well.name = "{}/{}".format(container["label"], well_data['name']) if well_data['name'] is not None else container["label"] well.properties = well_data['properties'] well.volume = Unit(well_data['volume_ul'], 'microliter') if 'ERROR' in well.properties: raise ValueError("Well {} has ERROR property: {}".format(well, well.properties["ERROR"])) if well.volume < Unit(20, "microliter"): logging.warn("Low volume for well {} : {}".format(well.name, well.volume)) return True def touchdown(fromC, toC, durations, stepsize=2, meltC=98, extC=72): """Touchdown PCR protocol generator""" assert 0 < stepsize < toC < fromC def td(temp, dur): return {"temperature":"{:2g}:celsius".format(temp), "duration":"{:d}:second".format(dur)} return [{"cycles": 1, "steps": [td(meltC, durations[0]), td(C, durations[1]), td(extC, durations[2])]} for C in np.arange(fromC, toC-stepsize, -stepsize)] def convert_ug_to_pmol(ug_dsDNA, num_nts): """Convert ug dsDNA to pmol""" return float(ug_dsDNA)/num_nts * (1e6 / 660.0) def expid(val): """Generate a unique ID per experiment""" return "{}_{}".format(experiment_name, val) def µl(microliters): """Unicode function name for creating microliter volumes""" return Unit(microliters,"microliter") DNA synthesis and synthetic biology
Despite the connection with modern synthetic biology, DNA synthesis is a rather old technology. For decades we have been able to make olikonucleotides (that is, DNA sequences up to 200 bases). However, it was always expensive, and chemistry never allowed for long DNA sequences. Recently it has become possible to synthesize whole genes (up to thousands of bases) at a reasonable price. This achievement really opens the era of "synthetic biology".
Craig Venter's Synthetic Genomics has advanced synthetic biology the furthest by synthesizing an entire body — over a million bases in length. As the length of the DNA increases, the problem becomes not synthesis, but assembly (i.e., stitching together the synthesized DNA sequences). With each assembly, you can double the length of DNA (or more), so after a dozen or so iterations a rather long molecule is obtained! The distinction between synthesis and assembly should soon become clear to the end user.
Moore's Law?
The price of DNA synthesis falls quite quickly, from more than $ 0.30 per basis two years ago to about $ 0.10 today, but it develops more like bacteria than processors. In contrast, DNA sequencing prices fall faster than Moore's law. A target of $ 0.02 per base is scheduled as an inflection point , where you can replace a lot of time consuming manipulations with DNA with simple synthesis. For example, at this price you can synthesize a whole plasmid of 3kb for $ 60 and skip a bunch of molecular biology. Hopefully we will achieve this in a couple of years.
Prices for DNA synthesis compared to prices for DNA sequencing, price for 1 basis (Carlson, 2014)
DNA synthesis companies
There are several large companies in the field of DNA synthesis: IDT is the largest producer of oliconucleotides, and can also produce longer (up to 2kb) “gene fragments” ( gBlocks ). Gen9 , Twist, and DNA 2.0 usually specialize in longer DNA sequences — these are gene synthesis companies. There are also some interesting new companies, such as Cambrian Genomics and Genesis DNA , which are working on the next generation synthesis methods.
Other companies, such as Amyris , Zymergen and Ginkgo Bioworks , use the DNA synthesized by these companies to work at the organism level. Synthetic Genomics also does this, but it itself synthesizes DNA.
Ginkgo recently made a deal with Twist to make 100 million bases: the biggest deal I saw. This proves that we live in the future, Twist even advertised the promotional code on Twitter: when you buy 10 million bases of DNA (almost the entire yeast genome!), You get another 10 million for free.
Twist Niche Suggestion on Twitter
Part One: Designing an Experiment
Green fluorescent protein
In this experiment, we synthesize a DNA sequence for a simple, green fluorescent protein (GFP). GFP protein was first found in a jellyfish that fluoresces under ultraviolet light. It is an extremely useful protein because it is easy to detect its expression by simply measuring the fluorescence. There are variations of GFP that produce yellow, red, orange, and other colors.
It is interesting to see how different mutations affect protein color, and this is a potentially interesting machine learning problem. Most recently, this would have to spend a lot of time in the lab, but now I will show you that it is (almost) as easy as editing a text file!
Technically, my GFP is a superfolder variant (sfGFP) with some mutations to improve qualities.
In superfolder-GFP (sfGFP), some mutations give it certain beneficial properties.
GFP structure (rendered using PV )
GFP Synthesis to Twist
I was lucky to get into the Twist alpha testing program, so I used their DNA synthesis service (they kindly placed my tiny order — thanks, Twist!). This is a new company in our field, with a new simplified synthesis process. Prices are around $ 0.10 for the base or lower , but they are still in beta , and the alpha program in which I participated has closed. Twist raised about $ 150 million, so their technology is lively enthusiastic.
I sent my DNA sequence to Twist as an Excel spreadsheet (there is no API yet, but I guess it will be soon), and they sent the synthesized DNA directly to my box at the Transcriptic lab (I also used IDT for synthesis, but they didn't send DNA right in Transcriptic, which spoils the fun a bit).
Obviously, this process has not yet become a typical use case and requires some support, but it worked, so the entire pipeline remains virtual. Without this, I probably would need access to the laboratory — many companies would not send DNA or reagents to their home address.
GFP is harmless, so any kind of light is highlighted
Plasmid vector
To express this protein in bacteria, the gene needs to live somewhere, otherwise the synthetic DNA encoding the gene simply instantly degrades. As a rule, in molecular biology we use a plasmid, a piece of circular DNA that lives outside the bacterial genome and expresses proteins. Plasmids are a convenient way for bacteria to share useful, autonomous functional modules, such as antibiotic resistance. There can be hundreds of plasmids in a cell.
A widely used terminology is that the plasmid is a vector , and synthetic DNA is an insertion. So here we are trying to clone the insertion into a vector, and then transform the bacteria with a vector.
Bacterial genome and plasmid (not to scale!) ( Wikipedia )
pUC19
I chose a fairly standard plasmid in the pUC19 series. This plasmid is very often used, and since it is available as part of the standard Transcriptic inventory, we do not need to send anything to them.
Structure of pUC19: the main components are the ampicillin resistance gene, lacZα, MCS / polylinker and the origin of replication (Wikipedia)
PUC19 has a pleasant function: since it contains the lacZα gene, you can use the blue-white selection method on it and see which colonies successfully inserted. Two chemicals are needed: IPTG and X-gal , and the scheme works as follows:
- IPTG induces the expression of lacZα.
- If lacZα is deactivated through DNA inserted into the multiple cloning site ( MCS / polylinker ) in lacZα, then the plasmid cannot hydrolyze X-gal, and these colonies will be white instead of blue.
- Therefore, a successful insertion produces white colonies, and a failed insertion produces blue colonies.
Blue-white selection shows where lacZα expression has been deactivated ( Wikipedia )
The openwetware documentation says:
E. coli DH5α does not require IPTG to induce expression from the lac promoter, even if the Lac repressor is expressed in the strain. The number of copies of most plasmids exceeds the number of repressors in the cells. If you need the maximum level of expression, add IPTG to a final concentration of 1 mM.
Synthetic DNA sequences
SfGFP DNA sequence
It is easy to obtain a DNA sequence for sfGFP by taking a protein sequence and encoding it with codons suitable for the host organism (here, E. coli ). This is a medium-sized protein with 236 amino acids, so at 10 cents per base, DNA synthesis costs about $ 70 .
Wolfram Alpha, the calculation of the cost of synthesis
The first 12 bases of our sfGFP is the Shine – Dalgarno sequence , which I added myself, which theoretically should increase the expression (AGGAGGACAGCT, then ATG ( start codon ) starts the protein). According to the computational tool developed by Salis Lab ( lecture slides ), we can expect medium to high expression of our protein (translation initiation rate is 10,000 "arbitrary units").
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641") Read in sfGFP plus Shine-Dalgarno: 726 bases long Translation matches protein with accession 532528641
DNA sequence of pUC19
First, I check that the sequence pUC19, which I downloaded from the NEB , is the correct length and includes the expected polylinker .
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker") Read in pUC19: 2686 bases long Found MCS / polylinker
We make some basic QC to make sure that EcoRI and BamHI are present in pUC19 only once (the following restriction enzymes are present in the Transcriptic inventory by default: PstI , PvuII , EcoRI , BamHI , BbsI , BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys())) Now we look at the lacZα sequence and check that there is nothing unexpected. For example, it must begin with Met and end with a stop codon. It is also easy to confirm that this is a full 324bp lacZα ORF by downloading the sequence pUC19 to the free snapgene viewer .
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence") lacZ sequence: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG r_MCS sequence: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC Found MCS once in lacZ sequence
Gibson assembly
DNA assembly simply means stitching the fragments. Usually you collect several DNA fragments into a longer segment, and then clone it into a plasmid or genome. In this experiment, I want to clone one DNA segment into plasmid pUC19 downstream of the lac promoter for expression in E. coli .
There are many ways to clone (for example, NEB , openwetware , addgene ). Here I will use the Gibson assembly ( developed by Daniel Gibson in Synthetic Genomics in 2009), which is not necessarily the cheapest method, but simple and flexible. You only need to place the DNA that you want to collect (with appropriate overlaps) in a tube with the Gibson Assembly Master Mix, and it is going to be on its own!
Gibson Assembly Review ( NEB )
Raw material
We start with 100 ng of synthetic DNA in 10 μl of liquid. This equals 0.21 picomoles of DNA or a concentration of 10 ng / μl.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp)) Insert: 100ng of DNA of length 726 equals 0.21 pmol
According to the NEB assembly protocol , this is enough source material:
NEB recommends a total of 0.02-0.5 picomoles of DNA fragments when 1 or 2 fragments are assembled into a vector, or 0.2-1.0 picomoles of DNA fragments when 4-6 fragments are assembled.
0.02-0.5 pmol * X μl
* Optimized cloning efficiency is 50-100 ng of vectors with a 2-3-fold excess of insertions. Use 5 times more inserts if the size is less than 200 bps. The total volume of unfiltered PCR fragments in a Gibson assembly reaction should not exceed 20%.
NEBuilder for Gibson assembly
Biolab's NEBuilder is a really great tool for creating a Gibson assembly protocol. PDF . pUC19 EcoRI, PCR [, — . .] .
:
:
- .
- .
- .
- .
1. PCR
The Gibson assembly depends on the DNA sequence that you are assembling, having some overlapping sequence (see the NEB protocol above for detailed instructions). In addition to simple amplification, PCR also allows you to add a flanking DNA sequence by simply inserting an additional sequence into the primers (you can also clone using only OE-PCR ).
We synthesize primers according to the NEB protocol above. I tried the Quickstart protocol on the Transcriptic website, but there is also an auto protocol command . Transcriptic , 1-2 ( , - , ).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"] IDT OligoAnalyzer . PCR primer dimer , NEB .
Gene-specific portion of flank (uppercase) Melt temperature: 51C, 53.5C Full sequence Melt temperature: 64.5C, 68.5C Hairpin: -.4dG, -5dG Self-dimer: -9dG, -16dG Heterodimer: -6dG
I went through many PCR iterations before getting satisfactory results, including experiments with several different brands of PCR mixes. Since each of these iterations can take several days (depending on the length of the queue to the laboratory), it is worthwhile to spend time in advance for debugging: it saves a lot of time in the long run. As the capacity of the cloud lab increases, this problem should become less acute. However, your first protocol is unlikely to succeed - there are too many variables.
Code
""" PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol() # --------------------------------------------------- # Set up experiment # experiment_name = "sfgfp_pcroe_v8" template_length = 740 _options = {'dilute_primers' : False, # if working stock has not been made 'dilute_template': False, # if working stock has not been made 'dilute_dNTP' : False, # if working stock has not been made 'run_gel' : True, # run a gel to see the plasmid size 'run_absorbance' : False, # check absorbance at 260/280/320 'run_sanger' : False} # sanger sequence the new sequence options = {k for k,v in _options.items() if v is True} # --------------------------------------------------- # Inventory and provisioning # https://developers.transcriptic.com/v1.0/docs/containers # # 'sfgfp2': 'ct17yx8h77tkme', # inventory; sfGFP tube #2, micro-1.5, cold_20 # 'sfgfp_puc19_primer1': 'ct17z9542mrcfv', # inventory; micro-2.0, cold_4 # 'sfgfp_puc19_primer2': 'ct17z9542m5ntb', # inventory; micro-2.0, cold_4 # 'sfgfp_idt_1ngul': 'ct184nnd3rbxfr', # inventory; micro-1.5, cold_4, (ERROR: no template) # inv = { 'Q5 Polymerase': 'rs16pcce8rdytv', # catalog; Q5 High-Fidelity DNA Polymerase 'Q5 Buffer': 'rs16pcce8rmke3', # catalog; Q5 Reaction Buffer 'dNTP Mixture': 'rs16pcb542c5rd', # catalog; dNTP Mixture (25mM?) 'water': 'rs17gmh5wafm5p', # catalog; Autoclaved MilliQ H2O 'sfgfp_pcroe_v5_puc19_primer1_10uM': 'ct186cj5cqzjmr', # inventory; micro-1.5, cold_4 'sfgfp_pcroe_v5_puc19_primer2_10uM': 'ct186cj5cq536x', # inventory; micro-1.5, cold_4 'sfgfp1': 'ct17yx8h759dk4', # inventory; sfGFP tube #1, micro-1.5, cold_20 } # Existing inventory template_tube = p.ref("sfgfp1", id=inv['sfgfp1'], cont_type="micro-1.5", storage="cold_4").well(0) dilute_primer_tubes = [p.ref('sfgfp_pcroe_v5_puc19_primer1_10uM', id=inv['sfgfp_pcroe_v5_puc19_primer1_10uM'], cont_type="micro-1.5", storage="cold_4").well(0), p.ref('sfgfp_pcroe_v5_puc19_primer2_10uM', id=inv['sfgfp_pcroe_v5_puc19_primer2_10uM'], cont_type="micro-1.5", storage="cold_4").well(0)] # New inventory resulting from this experiment dilute_template_tube = p.ref("sfgfp1_0.25ngul", cont_type="micro-1.5", storage="cold_4").well(0) dNTP_10uM_tube = p.ref("dNTP_10uM", cont_type="micro-1.5", storage="cold_4").well(0) sfgfp_pcroe_out_tube = p.ref(expid("amplified"), cont_type="micro-1.5", storage="cold_4").well(0) # Temporary tubes for use, then discarded mastermix_tube = p.ref("mastermix", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) water_tube = p.ref("water", cont_type="micro-1.5", storage="ambient", discard=True).well(0) pcr_plate = p.ref("pcr_plate", cont_type="96-pcr", storage="cold_4", discard=True) if 'run_absorbance' in options: abs_plate = p.ref("abs_plate", cont_type="96-flat", storage="cold_4", discard=True) # Initialize all existing inventory all_inventory_wells = [template_tube] + dilute_primer_tubes for well in all_inventory_wells: init_inventory_well(well) print(well.name, well.volume, well.properties) # ----------------------------------------------------- # Provision water once, for general use # p.provision(inv["water"], water_tube, µl(500)) # ----------------------------------------------------- # Dilute primers 1/10 (100uM->10uM) and keep at 4C # if 'dilute_primers' in options: for primer_num in (0,1): p.transfer(water_tube, dilute_primer_tubes[primer_num], µl(90)) p.transfer(primer_tubes[primer_num], dilute_primer_tubes[primer_num], µl(10), mix_before=True, mix_vol=µl(50)) p.mix(dilute_primer_tubes[primer_num], volume=µl(50), repetitions=10) # ----------------------------------------------------- # Dilute template 1/10 (10ng/ul->1ng/ul) and keep at 4C # OR # Dilute template 1/40 (10ng/ul->0.25ng/ul) and keep at 4C # if 'dilute_template' in options: p.transfer(water_tube, dilute_template_tube, µl(195)) p.mix(dilute_template_tube, volume=µl(100), repetitions=10) # Dilute dNTP to exactly 10uM if 'dilute_DNTP' in options: p.transfer(water_tube, dNTP_10uM_tube, µl(6)) p.provision(inv["dNTP Mixture"], dNTP_10uM_tube, µl(4)) # ----------------------------------------------------- # Q5 PCR protocol # www.neb.com/protocols/2013/12/13/pcr-using-q5-high-fidelity-dna-polymerase-m0491 # # 25ul reaction # ------------- # Q5 reaction buffer 5 µl # Q5 polymerase 0.25 µl # 10mM dNTP 0.5 µl -- 1µl = 4x12.5mM # 10uM primer 1 1.25 µl # 10uM primer 2 1.25 µl # 1pg-1ng Template 1 µl -- 0.5 or 1ng/ul concentration # ------------------------------- # Sum 9.25 µl # # # Mastermix tube will have 96ul of stuff, leaving space for 4x1ul aliquots of template p.transfer(water_tube, mastermix_tube, µl(64)) p.provision(inv["Q5 Buffer"], mastermix_tube, µl(20)) p.provision(inv['Q5 Polymerase'], mastermix_tube, µl(1)) p.transfer(dNTP_10uM_tube, mastermix_tube, µl(1), mix_before=True, mix_vol=µl(2)) p.transfer(dilute_primer_tubes[0], mastermix_tube, µl(5), mix_before=True, mix_vol=µl(10)) p.transfer(dilute_primer_tubes[1], mastermix_tube, µl(5), mix_before=True, mix_vol=µl(10)) p.mix(mastermix_tube, volume="48:microliter", repetitions=10) # Transfer mastermix to pcr_plate without template p.transfer(mastermix_tube, pcr_plate.wells(["A1","B1","C1"]), µl(24)) p.transfer(mastermix_tube, pcr_plate.wells(["A2"]), µl(24)) # acknowledged dead volume problems p.mix(pcr_plate.wells(["A1","B1","C1","A2"]), volume=µl(12), repetitions=10) # Finally add template p.transfer(template_tube, pcr_plate.wells(["A1","B1","C1"]), µl(1)) p.mix(pcr_plate.wells(["A1","B1","C1"]), volume=µl(12.5), repetitions=10) # --------------------------------------------------------- # Thermocycle with Q5 and hot start # 61.1 annealing temperature is recommended by NEB protocol # p.seal is enforced by transcriptic # extension_time = int(max(2, np.ceil(template_length * (11.0/1000)))) assert 0 < extension_time < 60, "extension time should be reasonable for PCR" cycles = [{"cycles": 1, "steps": [{"temperature": "98:celsius", "duration": "30:second"}]}] + \ touchdown(70, 61, [8, 25, extension_time], stepsize=0.5) + \ [{"cycles": 16, "steps": [{"temperature": "98:celsius", "duration": "8:second"}, {"temperature": "61.1:celsius", "duration": "25:second"}, {"temperature": "72:celsius", "duration": "{:d}:second".format(extension_time)}]}, {"cycles": 1, "steps": [{"temperature": "72:celsius", "duration": "2:minute"}]}] p.seal(pcr_plate) p.thermocycle(pcr_plate, cycles, volume=µl(25)) # -------------------------------------------------------- # Run a gel to hopefully see a 740bp fragment # if 'run_gel' in options: p.unseal(pcr_plate) p.mix(pcr_plate.wells(["A1","B1","C1","A2"]), volume=µl(12.5), repetitions=10) p.transfer(pcr_plate.wells(["A1","B1","C1","A2"]), pcr_plate.wells(["D1","E1","F1","D2"]), [µl(2), µl(4), µl(8), µl(8)]) p.transfer(water_tube, pcr_plate.wells(["D1","E1","F1","D2"]), [µl(18),µl(16),µl(12),µl(12)], mix_after=True, mix_vol=µl(10)) p.gel_separate(pcr_plate.wells(["D1","E1","F1","D2"]), µl(20), "agarose(10,2%)", "ladder1", "10:minute", expid("gel")) #--------------------------------------------------------- # Absorbance dilution series. Take 1ul out of the 25ul pcr plate wells # if 'run_absorbance' in options: p.unseal(pcr_plate) abs_wells = ["A1","B1","C1","A2","B2","C2","A3","B3","C3"] p.transfer(water_tube, abs_plate.wells(abs_wells[0:6]), µl(10)) p.transfer(water_tube, abs_plate.wells(abs_wells[6:9]), µl(9)) p.transfer(pcr_plate.wells(["A1","B1","C1"]), abs_plate.wells(["A1","B1","C1"]), µl(1), mix_after=True, mix_vol=µl(5)) p.transfer(abs_plate.wells(["A1","B1","C1"]), abs_plate.wells(["A2","B2","C2"]), µl(1), mix_after=True, mix_vol=µl(5)) p.transfer(abs_plate.wells(["A2","B2","C2"]), abs_plate.wells(["A3","B3","C3"]), µl(1), mix_after=True, mix_vol=µl(5)) for wavelength in [260, 280, 320]: p.absorbance(abs_plate, abs_plate.wells(abs_wells), "{}:nanometer".format(wavelength), exp_id("abs_{}".format(wavelength)), flashes=25) # ----------------------------------------------------------------------------- # Sanger sequencing: https://developers.transcriptic.com/docs/sanger-sequencing # "Each reaction should have a total volume of 15 µl and we recommend the following composition of DNA and primer: # PCR product (40 ng), primer (1 µl of a 10 µM stock)" # # By comparing to the gel ladder concentration (175ng/lane), it looks like 5ul of PCR product has approximately 30ng of DNA # if 'run_sanger' in options: p.unseal(pcr_plate) seq_wells = ["G1","G2"] for primer_num, seq_well in [(0, seq_wells[0]),(1, seq_wells[1])]: p.transfer(dilute_primer_tubes[primer_num], pcr_plate.wells([seq_well]), µl(1), mix_before=True, mix_vol=µl(50)) p.transfer(pcr_plate.wells(["A1"]), pcr_plate.wells([seq_well]), µl(5), mix_before=True, mix_vol=µl(10)) p.transfer(water_tube, pcr_plate.wells([seq_well]), µl(9)) p.mix(pcr_plate.wells(seq_wells), volume=µl(7.5), repetitions=10) p.sangerseq(pcr_plate, pcr_plate.wells(seq_wells[0]).indices(), expid("seq1")) p.sangerseq(pcr_plate, pcr_plate.wells(seq_wells[1]).indices(), expid("seq2")) # ------------------------------------------------------------------------- # Then consolidate to one tube. Leave at least 3ul dead volume in each tube # remaining_volumes = [well.volume - dead_volume['96-pcr'] for well in pcr_plate.wells(["A1","B1","C1"])] print("Consolidated volume", sum(remaining_volumes, µl(0))) p.consolidate(pcr_plate.wells(["A1","B1","C1"]), sfgfp_pcroe_out_tube, remaining_volumes, allow_carryover=True) uprint("\nProtocol 1. Amplify the insert (oligos previously synthesized)") jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) WARNING: root: Low volume for well sfGFP 1 / sfGFP 1: 2.0: microliter
sfGFP 1 / sfGFP 1 2.0: microliter {'dilution': '0.25ng / ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0: microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0: microliter {}
Consolidated volume 52.0: microliter
Protocol 1. Amplify the insert (oligos previously synthesized)
-------------------------------------------------- -------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
In the gel, you can estimate the correct size of the product after increasing the concentration (position of the strip in the gel) and the correct amount (darkness of the strip). The gel has a ladder corresponding to different lengths and quantities of DNA that can be used for comparison.
In the gel photograph below the band D1, E1, F1 contain, respectively, 2 μl, 4 μl and 8 μl of the amplified product. I can estimate the amount of DNA in each lane compared to DNA in the ladder (50 ng of DNA per lane in the ladder). I think the results look very clean.
I tried to use GelEval for image analysis and concentration assessment. , , , , . . GelEval 40 /.
, , dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
- EcoRI- pUC19, (D1, E1, F1), (D2)
PCR
Recently, Transcriptic began to provide interesting and useful diagnostic data from its robots. At the time of this writing, they are not available for download, so for now I only have an image of the temperatures during thermal cycling.
The data looks good, without unexpected peaks or valleys. A total of 35 PCR cycles, but some of these cycles are performed at a very high temperature within the PCR touchdown . In my previous attempts to amplify this segment - of which there were several! - there were problems with the hybridization of primers, so here PCR works a lot of time at high temperatures, which should improve the accuracy.
Thermocyclic diagnostics for a touchdown PCR: temperature block, sample and cover for 35 cycles and 42 minutes
Step 2. Cutting the plasmid
To insert our sfGFP DNA into pUC19, you first need to cut the plasmid. Following the NEB protocol, I do this using the EcoRI restriction enzyme . In the standard Transcriptic inventory there are reagents that I need: these are the NEB EcoRI and 10x CutSmart buffer , as well as the NEB pUC19 plasmid .
For information, lower prices from their inventory. In fact, I pay only part of the price, since Transcriptic takes payment for the quantity actually consumed:
Item ID Amount Concentration Price ------------ ------ ------------- ----------------- - ---- CutSmart 10x B7204S 5 ml 10 X $ 19.00 EcoRI R3101L 50,000 units 20,000 units / ml $ 225.00 pUC19 N3041L 250 µg 1,000 µg / ml $ 268.00
I followed the NEB protocol as much as possible:
. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
Code
"""Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p", # catalog; Autoclaved MilliQ H2O; ambient "pUC19": "rs17tcqmncjfsh", # catalog; pUC19; cold_20 "EcoRI": "rs17ta8xftpdk6", # catalog; EcoRI-HF; cold_20 "CutSmart": "rs17ta93g3y85t", # catalog; CutSmart Buffer 10x; cold_20 "ecori_p10x": "ct187v4ea85k2h", # inventory; EcoRI diluted 10x } # Tubes and plates I use then discard re_tube = p.ref("re_tube", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) water_tube = p.ref("water_tube", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) pcr_plate = p.ref("pcr_plate", cont_type="96-pcr", storage="cold_4", discard=True) # The result of the experiment, a pUC19 cut by EcoRI, goes in this tube for storage puc19_cut_tube = p.ref(expid("puc19_cut"), cont_type="micro-1.5", storage="cold_20").well(0) # ------------------------------------------------------------- # Provisioning and diluting. # Diluted EcoRI can be used more than once # p.provision(inv["water"], water_tube, µl(500)) if 'dilute_ecori' in options: ecori_p10x_tube = p.ref("ecori_p10x", cont_type="micro-1.5", storage="cold_20").well(0) p.transfer(water_tube, ecori_p10x_tube, µl(45)) p.provision(inv["EcoRI"], ecori_p10x_tube, µl(5)) else: # All "inventory" (stuff I own at transcriptic) must be initialized ecori_p10x_tube = p.ref("ecori_p10x", id=inv["ecori_p10x"], cont_type="micro-1.5", storage="cold_20").well(0) init_inventory_well(ecori_p10x_tube) # ------------------------------------------------------------- # Restriction enzyme cutting pUC19 # # 50ul total reaction volume for cutting 1ug of DNA: # 5ul CutSmart 10x # 1ul pUC19 (1ug of DNA) # 1ul EcoRI (or 10ul diluted EcoRI, 20 units, >10 units per ug DNA) # p.transfer(water_tube, re_tube, µl(117)) p.provision(inv["CutSmart"], re_tube, µl(15)) p.provision(inv["pUC19"], re_tube, µl(3)) p.mix(re_tube, volume=µl(60), repetitions=10) assert re_tube.volume == µl(120) + dead_volume["micro-1.5"] print("Volumes: re_tube:{} water_tube:{} EcoRI:{}".format(re_tube.volume, water_tube.volume, ecori_p10x_tube.volume)) p.distribute(re_tube, pcr_plate.wells(["A1","B1","A2"]), µl(40)) p.distribute(water_tube, pcr_plate.wells(["A2"]), µl(10)) p.distribute(ecori_p10x_tube, pcr_plate.wells(["A1","B1"]), µl(10)) assert all(well.volume == µl(50) for well in pcr_plate.wells(["A1","B1","A2"])) p.mix(pcr_plate.wells(["A1","B1","A2"]), volume=µl(25), repetitions=10) # Incubation to induce cut, then heat inactivation of EcoRI p.seal(pcr_plate) p.incubate(pcr_plate, "warm_37", "60:minute", shaking=False) p.thermocycle(pcr_plate, [{"cycles": 1, "steps": [{"temperature": "65:celsius", "duration": "21:minute"}]}], volume=µl(50)) # -------------------------------------------------------------- # Gel electrophoresis, to ensure the cutting worked # p.unseal(pcr_plate) p.mix(pcr_plate.wells(["A1","B1","A2"]), volume=µl(25), repetitions=5) p.transfer(pcr_plate.wells(["A1","B1","A2"]), pcr_plate.wells(["D1","E1","D2"]), µl(8)) p.transfer(water_tube, pcr_plate.wells(["D1","E1","D2"]), µl(15), mix_after=True, mix_vol=µl(10)) assert all(well.volume == µl(20) + dead_volume["96-pcr"] for well in pcr_plate.wells(["D1","E1","D2"])) p.gel_separate(pcr_plate.wells(["D1","E1","D2"]), µl(20), "agarose(10,2%)", "ladder2", "15:minute", expid("gel")) # ---------------------------------------------------------------------------- # Then consolidate all cut plasmid to one tube (puc19_cut_tube). # remaining_volumes = [well.volume - dead_volume['96-pcr'] for well in pcr_plate.wells(["A1","B1"])] print("Consolidated volume: {}".format(sum(remaining_volumes, µl(0)))) p.consolidate(pcr_plate.wells(["A1","B1"]), puc19_cut_tube, remaining_volumes, allow_carryover=True) assert all(tube.volume >= dead_volume['micro-1.5'] for tube in [water_tube, re_tube, puc19_cut_tube, ecori_p10x_tube]) # --------------------------------------------------------------- # Test protocol # jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze #print("Protocol {}\n\n{}".format(experiment_name, jprotocol)) open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter Consolidated volume: 78.0:microliter ✓ Protocol analyzed 12 instructions 5 containers Total Cost: $30.72 Workcell Time: $ 3.38 Reagents & Consumables: $ 27.34
Results: plasmid cutting
I conducted this experiment twice in slightly different conditions and with gels of different sizes, but the results are almost identical. I like both gels.
Initially, I did not allocate enough space for a “dead” volume (in test tubes 1.5 ml there is a dead volume of 15 μl!). I think this explains the difference between D1 and E1 (these two bands should be identical). The dead volume problem is easily solved by creating a proper working stock of diluted EcoRI at the beginning of the protocol.
Despite this error, in both gels the bands D1 and E1 are strong bands in the correct position 2,6kb. On the D2 band, uncut plasmid: as expected, it is not visible in one gel and is barely visible in another.
Two gel photos look quite different. This is partly due to the fact that Transcriptic has not yet automated this step.
Two gels showing cut pUC19 (2,6kb) on the D1 and E1 bands, and uncut pUC19 on D2
Step 3. Build by Gibson
, — , M13 ( ) qPCR , , . , , , .
, M13 , M13.
Code
"""Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p", # catalog; Autoclaved MilliQ H2O; ambient "M13_F" : "rs17tcpqwqcaxe", # catalog; M13 Forward (-41); cold_20 (1ul = 100pmol) "M13_R" : "rs17tcph6e2qzh", # catalog; M13 Reverse (-48); cold_20 (1ul = 100pmol) "SensiFAST_SYBR_No-ROX" : "rs17knkh7526ha", # catalog; SensiFAST SYBR for qPCR "sfgfp_puc19_gibson_v1_clone" : "ct187rzdq9kd7q", # inventory; assembled sfGFP; cold_4 "sfgfp_puc19_gibson_v3_clone" : "ct188ejywa8jcv", # inventory; assembled sfGFP; cold_4 } # --------------------------------------------------------------- # First get my sfGFP pUC19 clones, assembled with Gibson assembly # clone_plate1 = p.ref("sfgfp_puc19_gibson_v1_clone", id=inv["sfgfp_puc19_gibson_v1_clone"], cont_type="96-pcr", storage="cold_4", discard=False) clone_plate2 = p.ref("sfgfp_puc19_gibson_v3_clone", id=inv["sfgfp_puc19_gibson_v3_clone"], cont_type="96-pcr", storage="cold_4", discard=False) water_tube = p.ref("water", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) master_tube = p.ref("master", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) primer_tube = p.ref("primer", cont_type="micro-1.5", storage="cold_4", discard=True).well(0) pcr_plate = p.ref(expid("pcr_plate"), cont_type="96-pcr", storage="cold_4", discard=False) init_inventory_well(clone_plate1.well("A1")) init_inventory_well(clone_plate2.well("A1")) seq_wells = ["B2","B4","B6", # clone_plate1 "D2","D4","D6", # clone_plate2 "F2","F4"] # control # clone_plate2 was diluted 4X (20ul->80ul), according to NEB instructions assert clone_plate1.well("A1").volume == µl(18), clone_plate1.well("A1").volume assert clone_plate2.well("A1").volume == µl(78), clone_plate2.well("A1").volume # -------------------------------------------------------------- # Provisioning # p.provision(inv["water"], water_tube, µl(500)) # primers, diluted 2X, discarded at the end p.provision(inv["M13_F"], primer_tube, µl(13)) p.provision(inv["M13_R"], primer_tube, µl(13)) p.transfer(water_tube, primer_tube, µl(26), mix_after=True, mix_vol=µl(20), repetitions=10) # ------------------------------------------------------------------- # PCR Master mix -- 10ul SYBR mix, plus 1ul each undiluted primer DNA (100pmol) # Also add 15ul of dead volume # p.provision(inv['SensiFAST_SYBR_No-ROX'], master_tube, µl(11+len(seq_wells)*10)) p.transfer(primer_tube, master_tube, µl(4+len(seq_wells)*4)) p.mix(master_tube, volume=µl(63), repetitions=10) assert master_tube.volume == µl(127) # 15ul dead volume p.distribute(master_tube, pcr_plate.wells(seq_wells), µl(14), allow_carryover=True) p.distribute(water_tube, pcr_plate.wells(seq_wells), [µl(ul) for ul in [5,4,2, 4,2,0, 6,6]], allow_carryover=True) # Template -- starting with some small, unknown amount of DNA produced by Gibson p.transfer(clone_plate1.well("A1"), pcr_plate.wells(seq_wells[0:3]), [µl(1),µl(2),µl(4)], one_tip=True) p.transfer(clone_plate2.well("A1"), pcr_plate.wells(seq_wells[3:6]), [µl(2),µl(4),µl(6)], one_tip=True) assert all(pcr_plate.well(w).volume == µl(20) for w in seq_wells) assert clone_plate1.well("A1").volume == µl(11) assert clone_plate2.well("A1").volume == µl(66) # -------------------------------------------------------------- # qPCR # standard melting curve parameters # p.seal(pcr_plate) p.thermocycle(pcr_plate, [{"cycles": 1, "steps": [{"temperature": "95:celsius","duration": "2:minute"}]}, {"cycles": 40, "steps": [{"temperature": "95:celsius","duration": "5:second"}, {"temperature": "60:celsius","duration": "20:second"}, {"temperature": "72:celsius","duration": "15:second", "read": True}]}], volume=µl(20), # volume is optional dataref=expid("qpcr"), dyes={"SYBR": seq_wells}, # dye must be specified (tells transcriptic what aborbance to use?) melting_start="65:celsius", melting_end="95:celsius", melting_increment="0.5:celsius", melting_rate="5:second") # -------------------------------------------------------------- # Gel -- 20ul required # Dilute such that I have 11ul for sequencing # p.unseal(pcr_plate) p.distribute(water_tube, pcr_plate.wells(seq_wells), µl(11)) p.gel_separate(pcr_plate.wells(seq_wells), µl(20), "agarose(8,0.8%)", "ladder1", "10:minute", expid("gel")) # This appears to be a bug in Transcriptic. The actual volume should be 11ul # but it is not updating after running a gel with 20ul. # Primer tube should be equal to dead volume, or it's a waste assert all(pcr_plate.well(w).volume==µl(31) for w in seq_wells) assert primer_tube.volume == µl(16) == dead_volume['micro-1.5'] + µl(1) assert water_tube.volume > µl(25) # --------------------------------------------------------------- # Test and run protocol # jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) WARNING:root:Low volume for well sfgfp_puc19_gibson_v1_clone/sfgfp_puc19_gibson_v1_clone : 11.0:microliter
✓ Protocol analyzed 11 instructions 6 containers Total Cost: $32.09 Workcell Time: $ 6.98 Reagents & Consumables: $ 25.11
Results: qPCR for Gibson assembly
I can access qPCR data in JSON format via the Transcriptic API. This feature is not well documented , but can be extremely useful. The APIs even give you access to some diagnostic data from robots, which can aid in debugging.
First, we request the launch data:
project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json() Then we specify this id to get the qPCR “post-processing” data:
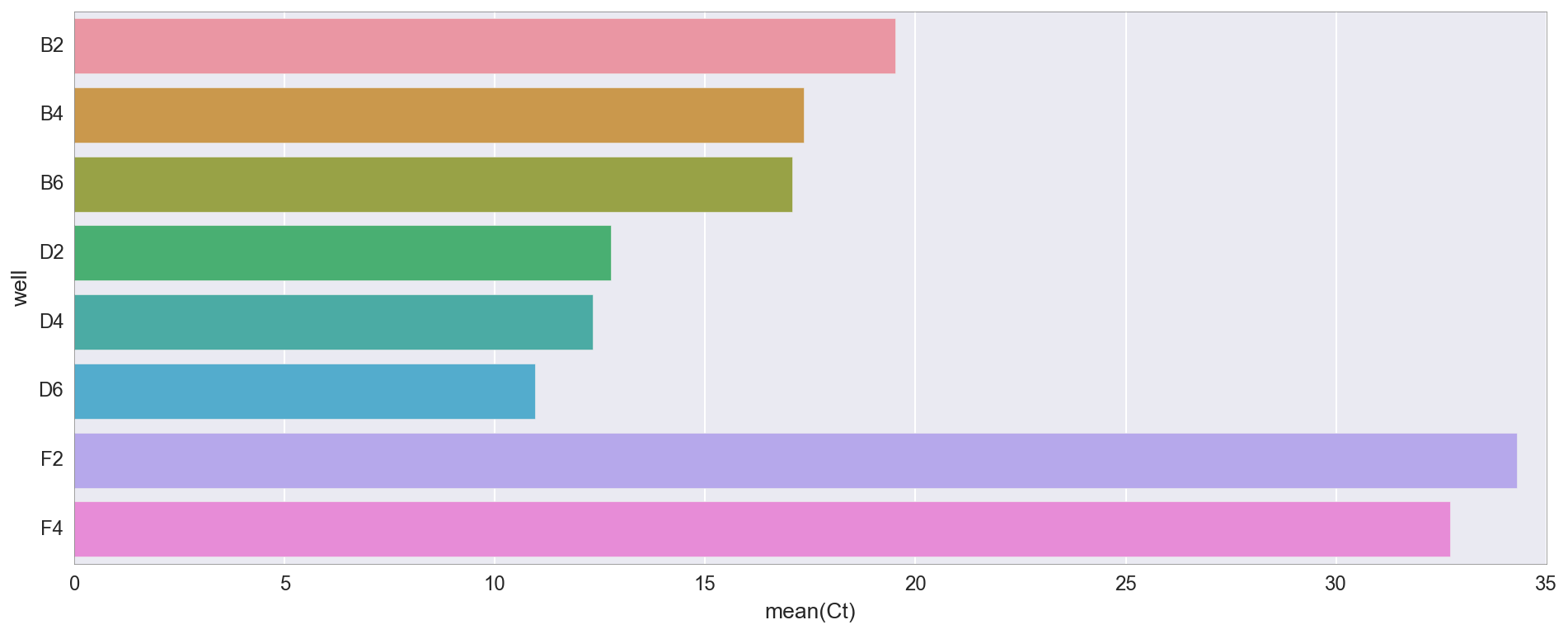
qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json() Here are the Ct values (cycle threshold) for each tube. Ct is just the point at which fluorescence exceeds a certain value. She roughly says how much DNA there is at the moment (and, therefore, approximately where we started).
# Simple util to convert wellnum to wellname n_w = {str(wellnum):'ABCDEFGH'[wellnum//12]+str(1+wellnum%12) for wellnum in range(96)} w_n = {v: k for k, v in n_w.items()} ct_vals = {n_w[k]:v for k,v in pp_data["amp0"]["SYBR"]["cts"].items()} ct_df = pd.DataFrame(ct_vals, index=["Ct"]).T ct_df["well"] = ct_df.index f, ax = plt.subplots(figsize=(16,6)) _ = sns.barplot(y="well", x="Ct", data=ct_df) , D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
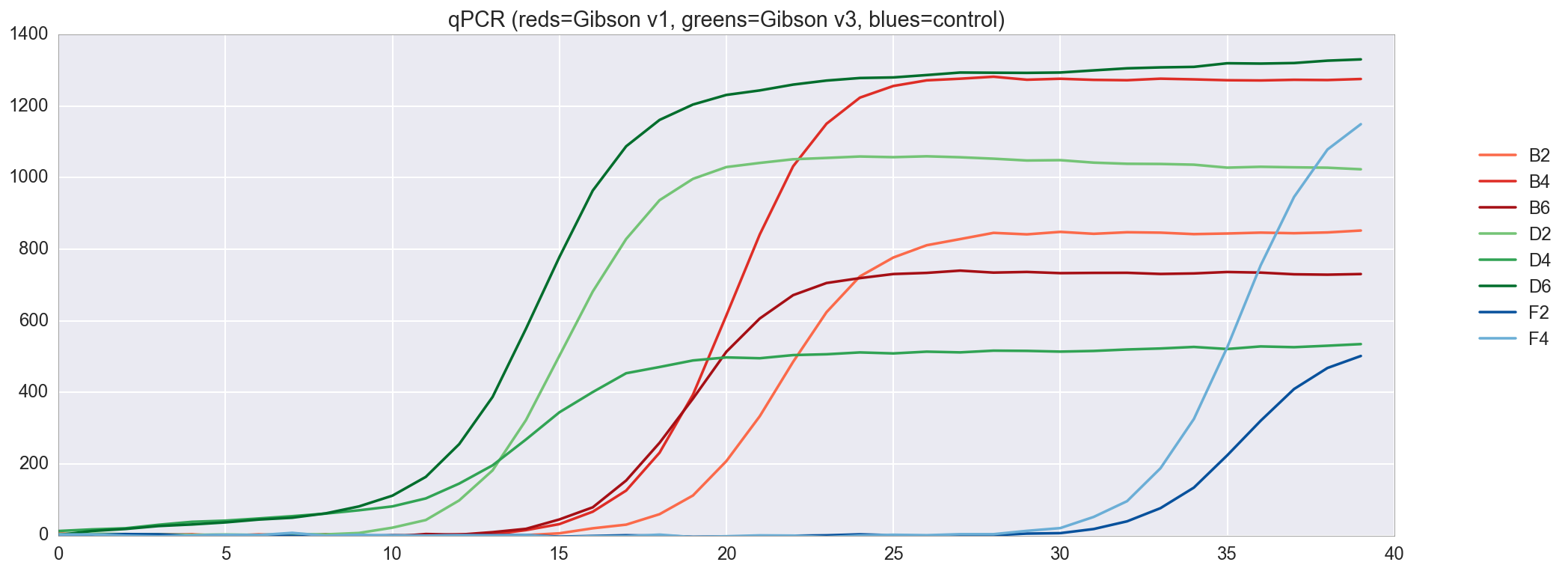
f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure) , qPCR , . v3 , v1, .
:
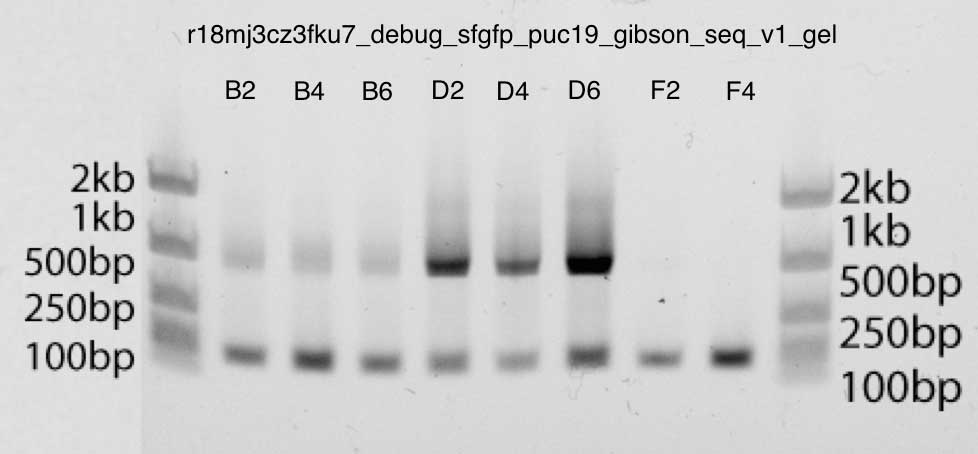
The gel is also very clean, shows strong bands just below 1kb in bands B2, B4, B6, D2, D4, D6: this is exactly the size we expect (insertion is about 740bp, and primers M13 are about 40bp up and down). The second bar corresponds to the primers. You can be sure of this, since the F2 and F4 bands contain only primer DNA.
Polyacrylamide gel electrophoresis: with the Gibson v3 assembly shows stronger bands (D2, D4, D6), in accordance with the qPCR data given above
Step 4. Transformation
Transformation is the process of changing the body by adding DNA. In this experiment, we transform E. coli using the sfGFP-expressing plasmid pUC19.
We use an easy-to-work strain Zymo DH5α Mix & Go and the recommended protocol . This strain is part of the standard Transcriptic inventory. In general, transformations can be complex, since competent cells are quite fragile, so the simpler and more reliable the protocol, the better. In ordinary molecular biology laboratories, these competent cells would probably be too expensive for general use.
Zymo Mix & Go cells with simple protocol
Problem with robots
This protocol is a good example of how difficult it is to adapt human protocols for use by robots and how it may unexpectedly fail. Protocols are sometimes surprisingly vague (“shake the tube from side to side”), based on the general context of molecular biologists, or they can suddenly request advanced image processing (“make sure the sediment is stirred”). People do not mind such tasks, but the robots need clearer instructions.
. , 37°C. , , , , Transcriptic — , . , , - , . . .
There are usually reasonable solutions: sometimes you just need to use different reagents (for example, more durable cells, such as Mix & Go above); sometimes you just lay actions with a margin (for example, shake ten times instead of three); Sometimes you need to come up with special tricks for robots (for example, use a PCR machine for heat stroke).
Of course, the big advantage is that, once the protocol has worked once, you can generally rely on it again and again. You can even quantify how reliable the protocol is and improve it over time!
Test transformation
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
Code
"""Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p", # catalog; Autoclaved MilliQ H2O; ambient "DH5a" : "rs16pbj944fnny", # catalog; Zymo DH5α; cold_80 "LB Miller" : "rs17bafcbmyrmh", # catalog; LB Broth Miller; cold_4 "Amp 100mgml" : "rs17msfk8ujkca", # catalog; Ampicillin 100mg/ml; cold_20 "pUC19" : "rs17tcqmncjfsh", # catalog; pUC19; cold_20 } # Catalog transform_plate = p.ref("transform_plate", cont_type="96-pcr", storage="ambient", discard=True) transform_tube = transform_plate.well(0) # ------------------------------------------------------------------------------------ # Plating transformed bacteria according to Tali's protocol (requires different code!) # http://learn.transcriptic.com/blog/2015/9/9/provisioning-commercial-reagents # Add 1-5ul plasmid and pre-warm culture plates to 37C before starting. # # # Extra inventory for plating # inv["lb-broth-100ug-ml-amp_6-flat"] = "ki17sbb845ssx9" # (kit, not normal ref) from blogpost inv["noAB-amp_6-flat"] = "ki17reefwqq3sq" # kit id inv["LB Miller"] = "rs17bafcbmyrmh" # # Ampicillin and no ampicillin plates # amp_6_flat = Container(None, p.container_type('6-flat')) p.refs["amp_6_flat"] = Ref('amp_6_flat', {"reserve": inv['lb-broth-100ug-ml-amp_6-flat'], "store": {"where": 'cold_4'}}, amp_6_flat) noAB_6_flat = Container(None, p.container_type('6-flat')) p.refs["noAB_6_flat"] = Ref('noAB_6_flat', {"reserve": inv['noAB-amp_6-flat'], "store": {"where": 'cold_4'}}, noAB_6_flat) # # Provision competent bacteria # p.provision(inv["DH5a"], transform_tube, µl(50)) p.provision(inv["pUC19"], transform_tube, µl(2)) # # Heatshock the bacteria to transform using a PCR machine # p.seal(transform_plate) p.thermocycle(transform_plate, [{"cycles": 1, "steps": [{"temperature": "4:celsius", "duration": "5:minute"}]}, {"cycles": 1, "steps": [{"temperature": "37:celsius", "duration": "30:minute"}]}], volume=µl(50)) p.unseal(transform_plate) # # Then dilute bacteria and spread onto 6-flat plates # Put more on ampicillin plates for more opportunities to get a colony # p.provision(inv["LB Miller"], transform_tube, µl(355)) p.mix(transform_tube, µl(150), repetitions=5) for i in range(6): p.spread(transform_tube, amp_6_flat.well(i), µl(55)) p.spread(transform_tube, noAB_6_flat.well(i), µl(10)) assert transform_tube.volume >= µl(15), transform_tube.volume # # Incubate and image 6-flat plates over 18 hours # for flat_name, flat in [("amp_6_flat", amp_6_flat), ("noAB_6_flat", noAB_6_flat)]: for timepoint in [6,12,18]: p.cover(flat) p.incubate(flat, "warm_37", "6:hour") p.uncover(flat) p.image_plate(flat, mode="top", dataref=expid("{}_t{}".format(flat_name, timepoint))) # --------------------------------------------------------------- # Analyze protocol # jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze #print("Protocol {}\n\n{}".format(experiment_name, protocol)) open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) ✓ Protocol analyzed 43 instructions 3 containers $45.43
:
In the following photos, we see that without the antibiotic (the plate on the left), growth is observed on all six plates, although strongly to varying degrees, which causes concern. It seems that Transcriptic robots do not really cope with the uniform distribution, which requires some dexterity.
( ) , . , , , , 55 10 . . , . , , .
( , , , E. coli . Growth is much weaker on plates with ampicillin, although there are much more bacteria, as expected).
In general, the transformation worked well enough to continue, although there are some flaws.
Plates of cells transformed with pUC19, after 18 hours: without antibiotic (left) and with antibiotic (right)
Product Transformation After Assembly
Since the Gibson assembly and simple transformation of pUC19 seem to work, you can now try the transformation with a fully assembled plasmid expressing sfGFP.
In addition to the assembled insertion, I will also add some IPTG and X-gal to the plates to see a successful transformation using the blue-white selection method . This additional information is useful, because if a transformation with the usual pUC19, which does not contain sfGFP, goes through, it will still give resistance to antibiotics.
Absorption and fluorescence
According to this table , sfGFP shines best at excitation wavelengths of 485 nm / 510 nm. I found that 485/535 works better in Transcriptic. I guess because 485 and 510 are too similar. I measure bacterial growth at 600 nm ( OD600 ).
A variety of GFP ( biotek )
IPTG and X-gal
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
Code
"""Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False, # do a new gibson assembly "sanger" : False, # sanger sequence product "control_pUC19" : True, # unassembled pUC19 "XGal" : False # excluding X-gal should make the colony picking easier } for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_plates_v4" # ----------------------------------------------------------------------- # Inventory # inv = { # catalog "water" : "rs17gmh5wafm5p", # catalog; Autoclaved MilliQ H2O; ambient "DH5a" : "rs16pbj944fnny", # catalog; Zymo DH5α; cold_80 "Gibson Mix" : "rs16pfatkggmk5", # catalog; Gibson Mix (2X); cold_20 "LB Miller" : "rs17bafcbmyrmh", # catalog; LB Broth Miller; cold_4 "Amp 100mgml" : "rs17msfk8ujkca", # catalog; Ampicillin 100mg/ml; cold_20 "pUC19" : "rs17tcqmncjfsh", # catalog; pUC19; cold_20 # my inventory "puc19_cut_v2": "ct187v4ea7vvca", # inventory; pUC19 cut with EcoRI; cold_20 "IPTG" : "ct18a2r5wn6tqz", # inventory; IPTG at 1M (conc semi-documented); cold_20 "XGal" : "ct18a2r5wp5hcv", # inventory; XGal at 0.1M (conc not documented); cold_20 "sfgfp_pcroe_v8_amplified" : "ct1874zqh22pab", # inventory; sfGFP amplified to 40ng/ul; cold_4 "sfgfp_puc19_gibson_v3_clone" : "ct188ejywa8jcv", # inventory; assembled sfGFP; cold_4 # kits (must be used differently) "lb-broth-100ug-ml-amp_6-flat" : "ki17sbb845ssx9", # catalog; ampicillin plates "noAB-amp_6-flat" : "ki17reefwqq3sq" # catalog; no antibiotic plates } # # Catalog (all to be discarded afterward) # water_tube = p.ref("water", cont_type="micro-1.5", storage="ambient", discard=True).well(0) transform_plate = p.ref("trn_plate", cont_type="96-pcr", storage="ambient", discard=True) transform_tube = transform_plate.well(39) # experiment transform_tube_L = p.ref("trn_tubeL", cont_type="micro-1.5", storage="ambient", discard=True).well(0) transctrl_tube = transform_plate.well(56) # control transctrl_tube_L = p.ref("trc_tubeL", cont_type="micro-1.5", storage="ambient", discard=True).well(0) # # Plating according to Tali's protocol # http://learn.transcriptic.com/blog/2015/9/9/provisioning-commercial-reagents # amp_6_flat = Container(None, p.container_type('6-flat')) p.refs[expid("amp_6_flat")] = Ref(expid("amp_6_flat"), {"reserve": inv['lb-broth-100ug-ml-amp_6-flat'], "store": {"where": 'cold_4'}}, amp_6_flat) noAB_6_flat = Container(None, p.container_type('6-flat')) p.refs[expid("noAB_6_flat")] = Ref(expid("noAB_6_flat"), {"reserve": inv['noAB-amp_6-flat'], "store": {"where": 'cold_4'}}, noAB_6_flat) # # My inventory: EcoRI-cut pUC19, oePCR'd sfGFP, Gibson-assembled pUC19, IPTG and X-Gal # if "gibson" in options: puc19_cut_tube = p.ref("puc19_ecori_v2_puc19_cut", id=inv["puc19_cut_v2"], cont_type="micro-1.5", storage="cold_20").well(0) sfgfp_pcroe_amp_tube = p.ref("sfgfp_pcroe_v8_amplified", id=inv["sfgfp_pcroe_v8_amplified"], cont_type="micro-1.5", storage="cold_4").well(0) clone_plate = p.ref(expid("clone"), cont_type="96-pcr", storage="cold_4", discard=False) else: clone_plate = p.ref("sfgfp_puc19_gibson_v3_clone", id=inv["sfgfp_puc19_gibson_v3_clone"], cont_type="96-pcr", storage="cold_4", discard=False) IPTG_tube = p.ref("IPTG", id=inv["IPTG"], cont_type="micro-1.5", storage="cold_20").well(0) if "XGal" in options: XGal_tube = p.ref("XGal", id=inv["XGal"], cont_type="micro-1.5", storage="cold_20").well(0) # # Initialize inventory # if "gibson" in options: all_inventory_wells = [puc19_cut_tube, sfgfp_pcroe_amp_tube, IPTG_tube] assert puc19_cut_tube.volume == µl(66), puc19_cut_tube.volume assert sfgfp_pcroe_amp_tube.volume == µl(36), sfgfp_pcroe_amp_tube.volume else: all_inventory_wells = [IPTG_tube, clone_plate.well(0)] if "XGal" in options: all_inventory_wells.append(XGal_tube) for well in all_inventory_wells: init_inventory_well(well) print("Inventory: {} {} {}".format(well.name, well.volume, well.properties)) # # Provisioning. Water is used all over the protocol. Provision an excess since it's cheap # p.provision(inv["water"], water_tube, µl(500)) # ----------------------------------------------------------------------------- # Cloning/assembly (see NEBuilder protocol above) # # "Optimized efficiency is 50–100 ng of vectors with 2 fold excess of inserts." # pUC19 is 20ng/ul (78ul total). # sfGFP is ~40ng/ul (48ul total) # Therefore 4ul of each gives 80ng and 160ng of vector and insert respectively # def do_gibson_assembly(): # # Combine all the Gibson reagents in one tube and thermocycle # p.provision(inv["Gibson Mix"], clone_plate.well(0), µl(10)) p.transfer(water_tube, clone_plate.well(0), µl(2)) p.transfer(puc19_cut_tube, clone_plate.well(0), µl(4)) p.transfer(sfgfp_pcroe_amp_tube, clone_plate.well(0), µl(4), mix_after=True, mix_vol=µl(10), repetitions=10) p.seal(clone_plate) p.thermocycle(clone_plate, [{"cycles": 1, "steps": [{"temperature": "50:celsius", "duration": "16:minute"}]}], volume=µl(50)) # # Dilute assembled plasmid 4X according to the NEB Gibson assembly protocol (20ul->80ul) # p.unseal(clone_plate) p.transfer(water_tube, clone_plate.well(0), µl(60), mix_after=True, mix_vol=µl(40), repetitions=5) return # -------------------------------------------------------------------------------------------------- # Transformation # "Transform NEB 5-alpha Competent E. coli cells with 2 μl of the # assembled product, following the appropriate transformation protocol." # # Mix & Go http://www.zymoresearch.com/downloads/dl/file/id/173/t3015i.pdf # "[After mixing] Immediately place on ice and incubate for 2-5 minutes" # "The highest transformation efficiencies can be obtained by incubating Mix & Go cells with DNA on # ice for 2-5 minutes (60 minutes maximum) prior to plating." # "It is recommended that culture plates be pre-warmed to >20°C (preferably 37°C) prior to plating." # "Avoid exposing the cells to room temperature for more than a few seconds at a time." # # "If competent cells are purchased from other manufacture, dilute assembled products 4-fold # with H2O prior transformation. This can be achieved by mixing 5 μl of assembled products with # 15 μl of H2O. Add 2 μl of the diluted assembled product to competent cells." # def _do_transformation(): # # Combine plasmid and competent bacteria in a pcr_plate and shock # p.provision(inv["DH5a"], transform_tube, µl(50)) p.transfer(clone_plate.well(0), transform_tube, µl(3), dispense_speed="10:microliter/second") assert clone_plate.well(0).volume == µl(54), clone_plate.well(0).volume if 'control_pUC19' in options: p.provision(inv["DH5a"], transctrl_tube, µl(50)) p.provision(inv["pUC19"], transctrl_tube, µl(1)) # # Heatshock the bacteria to transform using a PCR machine # p.seal(transform_plate) p.thermocycle(transform_plate, [{"cycles": 1, "steps": [{"temperature": "4:celsius", "duration": "5:minute"}]}, {"cycles": 1, "steps": [{"temperature": "37:celsius", "duration": "30:minute"}]}], volume=µl(50)) return def _transfer_transformed_to_plates(): assert transform_tube.volume == µl(53), transform_tube.volume p.unseal(transform_plate) num_ab_plates = 4 # antibiotic places # # Transfer bacteria to a bigger tube for diluting # Then spread onto 6-flat plates # Generally you would spread 50-100ul of diluted bacteria # Put more on ampicillin plates for more opportunities to get a colony # I use a dilution series since it's unclear how much to plate # p.provision(inv["LB Miller"], transform_tube_L, µl(429)) # # Add all IPTG and XGal to the master tube # 4ul (1M) IPTG on each plate; 40ul XGal on each plate # p.transfer(IPTG_tube, transform_tube_L, µl(4*num_ab_plates)) if 'XGal' in options: p.transfer(XGal_tube, transform_tube_L, µl(40*num_ab_plates)) # # Add the transformed cells and mix (use new mix op in case of different pipette) # p.transfer(transform_tube, transform_tube_L, µl(50)) p.mix(transform_tube_L, volume=transform_tube_L.volume/2, repetitions=10) assert transform_tube.volume == dead_volume['96-pcr'] == µl(3), transform_tube.volume assert transform_tube_L.volume == µl(495), transform_tube_L.volume # # Spread an average of 60ul on each plate == 480ul total # for i in range(num_ab_plates): p.spread(transform_tube_L, amp_6_flat.well(i), µl(51+i*6)) p.spread(transform_tube_L, noAB_6_flat.well(i), µl(51+i*6)) assert transform_tube_L.volume == dead_volume["micro-1.5"], transform_tube_L.volume # # Controls: include 2 ordinary pUC19-transformed plates as a control # if 'control_pUC19' in options: num_ctrl = 2 assert num_ab_plates + num_ctrl <= 6 p.provision(inv["LB Miller"], transctrl_tube_L, µl(184)+dead_volume["micro-1.5"]) p.transfer(IPTG_tube, transctrl_tube_L, µl(4*num_ctrl)) if "XGal" in options: p.transfer(XGal_tube, transctrl_tube_L, µl(40*num_ctrl)) p.transfer(transctrl_tube, transctrl_tube_L, µl(48)) p.mix(transctrl_tube_L, volume=transctrl_tube_L.volume/2, repetitions=10) for i in range(num_ctrl): p.spread(transctrl_tube_L, amp_6_flat.well(num_ab_plates+i), µl(55+i*10)) p.spread(transctrl_tube_L, noAB_6_flat.well(num_ab_plates+i), µl(55+i*10)) assert transctrl_tube_L.volume == dead_volume["micro-1.5"], transctrl_tube_L.volume assert IPTG_tube.volume == µl(808), IPTG_tube.volume if "XGal" in options: assert XGal_tube.volume == µl(516), XGal_tube.volume return def do_transformation(): _do_transformation() _transfer_transformed_to_plates() # ------------------------------------------------------ # Measure growth in plates (photograph) # def measure_growth(): # # Incubate and photograph 6-flat plates over 18 hours # to see blue or white colonies # for flat_name, flat in [(expid("amp_6_flat"), amp_6_flat), (expid("noAB_6_flat"), noAB_6_flat)]: for timepoint in [9,18]: p.cover(flat) p.incubate(flat, "warm_37", "9:hour") p.uncover(flat) p.image_plate(flat, mode="top", dataref=expid("{}_t{}".format(flat_name, timepoint))) return # --------------------------------------------------------------- # Sanger sequencing, TURNED OFF # Sequence to make sure assembly worked # 500ng plasmid, 1 µl of a 10 µM stock primer # "M13_F" : "rs17tcpqwqcaxe", # catalog; M13 Forward (-41); cold_20 (1ul = 100pmol) # "M13_R" : "rs17tcph6e2qzh", # catalog; M13 Reverse (-48); cold_20 (1ul = 100pmol) # def do_sanger_seq(): seq_primers = [inv["M13_F"], inv["M13_R"]] seq_wells = ["G1","G2"] p.unseal(pcr_plate) for primer_num, seq_well in [(0, seq_wells[0]),(1, seq_wells[1])]: p.provision(seq_primers[primer_num], pcr_plate.wells([seq_well]), µl(1)) p.transfer(pcr_plate.wells(["A1"]), pcr_plate.wells(seq_wells), µl(5), mix_before=True, mix_vol=µl(10)) p.transfer(water_tube, pcr_plate.wells(seq_wells), µl(9)) p.mix(pcr_plate.wells(seq_wells), volume=µl(7.5), repetitions=10) p.sangerseq(pcr_plate, pcr_plate.wells(seq_wells[0]).indices(), expid("seq1")) p.sangerseq(pcr_plate, pcr_plate.wells(seq_wells[1]).indices(), expid("seq2")) return # --------------------------------------------------------------- # Generate protocol # # Skip Gibson since I already did it if 'gibson' in options: do_gibson_assembly() do_transformation() measure_growth() if 'sanger' in options: do_sanger_seq() # --------------------------------------------------------------- # Output protocol # jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze #print("\nProtocol {}\n\n{}".format(experiment_name, jprotocol)) open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) Inventory: IPTG/IPTG/IPTG/IPTG/IPTG/IPTG 832.0:microliter {}
Inventory: sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone 57.0:microliter {}
✓ Protocol analyzed
40 instructions
8 containers
Total Cost: $53.20
Workcell Time: $ 17.35
Reagents & Consumables: $ 35.86 Collecting the colonies
When colonies grow on an ampicillin plate, I can “collect” individual colonies and plant them on a 96-tube plate. For this, there is a special command ( autopick ) in the autoprotocol .
Code
"""Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val) # ----------------------------------------------------------------------- # Inventory # inv = { # catalog "water" : "rs17gmh5wafm5p", # catalog; Autoclaved MilliQ H2O; ambient "LB Miller" : "rs17bafcbmyrmh", # catalog; LB Broth Miller; cold_4 "Amp 100mgml" : "rs17msfk8ujkca", # catalog; Ampicillin 100mg/ml; cold_20 "IPTG" : "ct18a2r5wn6tqz", # inventory; IPTG at 1M (conc semi-documented); cold_20 # plates from previous experiment, must be changed every new experiment plate_expid("amp_6_flat") : "ct18snmr9avvg9", # inventory; Ampicillin plates with blue-white screening of pUC19 plate_expid("noAB_6_flat") : "ct18snmr9dxfw2", # inventory; no AB plates with blue-white screening of pUC19 } # Tubes and plates lb_amp_tubes = [p.ref("lb_amp_{}".format(i+1), cont_type="micro-2.0", storage="ambient", discard=True).well(0) for i in range(4)] lb_xab_tube = p.ref("lb_xab", cont_type="micro-2.0", storage="ambient", discard=True).well(0) growth_plate = p.ref(expid("growth"), cont_type="96-flat", storage="cold_4", discard=False) # My inventory IPTG_tube = p.ref("IPTG", id=inv["IPTG"], cont_type="micro-1.5", storage="cold_20").well(0) # ampicillin plate amp_6_flat = Container(None, p.container_type('6-flat')) p.refs[plate_expid("amp_6_flat")] = Ref(plate_expid("amp_6_flat"), {"id":inv[plate_expid("amp_6_flat")], "store": {"where": 'cold_4'}}, amp_6_flat) # Use a total of 50 wells abs_wells = ["{}{}".format(row,col) for row in "BCDEF" for col in range(1,11)] abs_wells_T = ["{}{}".format(row,col) for col in range(1,11) for row in "BCDEF"] assert abs_wells[:3] == ["B1","B2","B3"] and abs_wells_T[:3] == ["B1","C1","D1"] def prepare_growth_wells(): # # To LB, add ampicillin at ~1/1000 concentration # Mix slowly in case of overflow # p.provision(inv["LB Miller"], lb_xab_tube, µl(1913)) for lb_amp_tube in lb_amp_tubes: p.provision(inv["Amp 100mgml"], lb_amp_tube, µl(2)) p.provision(inv["LB Miller"], lb_amp_tube, µl(1911)) p.mix(lb_amp_tube, volume=µl(800), repetitions=10) # # Add IPTG but save on X-Gal # http://openwetware.org/images/f/f1/Dh5a_sub.pdf # "If you are concerned about obtaining maximal levels of expression, add IPTG to a final concentration of 1 mM." # 2ul of IPTG in 2000ul equals 1mM # p.transfer(IPTG_tube, [lb_xab_tube] + lb_amp_tubes, µl(2), one_tip=True) # # Distribute LB among wells, row D is control (no ampicillin) # cols = range(1,11) row = "D" # control, no AB cwells = ["{}{}".format(row,col) for col in cols] assert set(cwells).issubset(set(abs_wells)) p.distribute(lb_xab_tube, growth_plate.wells(cwells), µl(190), allow_carryover=True) rows = "BCEF" for row, lb_amp_tube in zip(rows, lb_amp_tubes): cwells = ["{}{}".format(row,col) for col in cols] assert set(cwells).issubset(set(abs_wells)) p.distribute(lb_amp_tube, growth_plate.wells(cwells), µl(190), allow_carryover=True) assert all(lb_amp_tube.volume == lb_xab_tube.volume == dead_volume['micro-2.0'] for lb_amp_tube in lb_amp_tubes) return def measure_growth_wells(): # # Growth: absorbance and fluorescence over 24 hours # Absorbance at 600nm: cell growth # Absorbance at 615nm: X-gal, in theory # Fluorescence at 485nm/510nm: sfGFP # or 450nm/508nm (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2695656/) # hr = 4 for t in range(0,24,hr): if t > 0: p.cover(growth_plate) p.incubate(growth_plate, "warm_37", "{}:hour".format(hr), shaking=True) p.uncover(growth_plate) p.fluorescence(growth_plate, growth_plate.wells(abs_wells).indices(), excitation="485:nanometer", emission="535:nanometer", dataref=expid("fl2_{}".format(t)), flashes=25) p.fluorescence(growth_plate, growth_plate.wells(abs_wells).indices(), excitation="450:nanometer", emission="508:nanometer", dataref=expid("fl1_{}".format(t)), flashes=25) p.fluorescence(growth_plate, growth_plate.wells(abs_wells).indices(), excitation="395:nanometer", emission="508:nanometer", dataref=expid("fl0_{}".format(t)), flashes=25) p.absorbance(growth_plate, growth_plate.wells(abs_wells).indices(), wavelength="600:nanometer", dataref=expid("abs_{}".format(t)), flashes=25) return # --------------------------------------------------------------- # Protocol steps # prepare_growth_wells() batch = 10 for i in range(5): p.autopick(amp_6_flat.well(i), growth_plate.wells(abs_wells_T[i*batch:i*batch+batch]), dataref=expid("autopick_{}".format(i))) p.image_plate(amp_6_flat, mode="top", dataref=expid("autopicked_{}".format(i))) measure_growth_wells() # --------------------------------------------------------------- # Output protocol # jprotocol = json.dumps(p.as_dict(), indent=2) !echo '{jprotocol}' | transcriptic analyze open("protocol_{}.json".format(experiment_name),'w').write(jprotocol) ✓ Protocol analyzed 62 instructions 8 containers Total Cost: $ 66.38 Workcell Time: $ 57.59 Reagents & Consumables: $ 8.78
Results: collection of colonies
The blue-white screen perfectly showed mainly white colonies on plates with antibiotics (1-4) and only blue ones on plates without antibiotics (5-6). This is exactly what I expected, and I was glad to see it, especially since I used my own IPTG and X-gal, which I sent to Transcriptic.
Screening using white-blue plate selection with ampicillin (1-4) and without antibiotic (5-6)
However, the robot-collector of colonies did not work well with these white-blue colonies. The image below is created by subtracting consecutive photographs of the plates after each round of plate selection and increasing the contrast of the differences (in GraphicsMagick ). In this way, I can visualize which colonies were collected (although not perfect, because the collected colonies are not completely removed).
I also signed the image with the number of colonies collected by the Transcriptic robot. It was assumed that he will collect a maximum of 10 colonies from the first five plates. However, several colonies were generally collected, and these are usually blue colonies. The robot only managed to find ten colonies on a control plate with only blue colonies. My working theory is that a robot collecting colonies preferably collects blue colonies, since they are more contrasting.
Screening plates for white-blue breeding with ampicillin (1-4) and without antibiotic (5-6), indicating the number of collected colonies
The blue-white screening served a specific purpose. He showed that most colonies are transformed correctly. At least there is an insertion. However, for a better collection of colonies, I repeated the experiment without X-gal.
Only with white colonies, the robot assembler successfully collected ten colonies from each of the first five plates. It can be assumed that in most of the collected colonies there are successful insertions.
Colonies growing on plates with ampicillin (1-4) and without antibiotic (5-6)
Results: transformation with the collected product
After growing 50 selected colonies on a 96-well plate for 20 hours, I measure the fluorescence to check for sfGFP expression. Transcriptic uses a Tecan Infinite reader to measure fluorescence and absorption (and luminescence, if you want) .
In theory, an assembled plasmid should be collected in any colony with growth, since it needs antibiotic resistance for growth, and each assembled plasmid expresses sfGFP. In fact, there are many reasons why this may not be the case, not least because you can lose the sfGFP gene from a plasmid without losing resistance to ampicillin. A bacterium that loses the sfGFP gene has an advantage in selection over its competitors, because it does not waste excess energy, and with a sufficient number of generations of growth, this will definitely happen.
I collect absorption (OD600) and fluorescence data every four hours for 20 hours (about 60 generations).
for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:]) We place on the chart the data of the 20th hour and the traces of previous measurements. Actually, I am only interested in the latest data, since that is when the peak of fluorescence should be observed.
svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()] # axes svg.append('<g fill="#888" font-size="18" transform="translate(20,0),scale(.95)">') svg.append('<text x="0" y="{}">OD600 →</text>'.format(H+20)) svg.append('<text x="0" y="0" transform="rotate(-90),translate(-{},-8)">Fluorescence →</text>'.format(H)) svg.append('<line x1="0" y1="{}" x2="{}" y2="{}" style="stroke:#888;stroke-width:2" />'.format(H,W,H)) svg.append('<line x1="0" y1="0" x2="0" y2="{}" style="stroke:#888;stroke-width:2" />'.format(H)) # glow filter svg.append("""<filter id="glow" x="-200%" y="-200%" height="400%" width="400%"> <feColorMatrix type="matrix" values="0 0 0 0 0 255 0 0 0 0 0 0 0 0 0 0 0 0 1 0"/> <feGaussianBlur stdDeviation="10" result="coloredBlur"/> <feMerge><feMergeNode in="coloredBlur"/><feMergeNode in="SourceGraphic"/></feMerge> </filter>""") for n, (well, vals) in enumerate(ab_fls): fill = "#444" if not well.startswith("D") else "#aaa" gfilter = 'filter="url(#glow)"' if well in ["C3", "D1", "D3"] else "" cx, cy = _toxy(*vals[-2:]) svg.append('''<g id="point{n:d}"><circle {gfilter:s} r="12" cx="{cx:f}" cy="{cy:f}" fill="{fill:s}" /> <text x="{cx:f}" y="{cy:f}" font-size="10" text-anchor="middle" fill="#fff" alignment-baseline="middle">{txt:s}</text></g> '''.format(n=n, cx=cx, cy=cy, fill=fill, txt=well, gfilter=gfilter)) pathd = 'M{} '.format(_topt(*vals[:2])) pathd += ' '.join("L{}".format(_topt(*vals[i:i+2])) for i in range(2,len(vals),2)) svg.append('''<path d="{pathd:}" stroke="#ccc" stroke-width=".2" fill="none" id="path{n:d}"/>'''.format(pathd=pathd, n=n)) svg.append("</g>") # entire chart group show_svg(''.join(svg), w=W, h=H) OD600: , . , sfGFP
miniprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
Based on fluorescence data and sequencing results, it appears that only three of the 50 colonies produce sfGFP and fluoresce. This is not as much as I expected. However, since three separate growth stages have passed (on a plate, in a test tube, for miniprep), about 200 generations of growth have undergone to this stage, so there were quite a few possibilities for mutations to occur.
There must be ways to make the process more efficient, especially since I am far from being an expert on these protocols. However, we successfully produced transformed cells with the expression of engineered GFP using only Python code!
Part Three: Conclusions
Price
Depending on how to measure, the cost of this experiment was about $ 360, not counting money for debugging:
- $ 70 for DNA synthesis
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
I think that the cost can be reduced to $ 250-300 with some modifications. For example, a robotic collection of 50 colonies is suspiciously expensive and can probably be abandoned.
In my experience, this price seems high for some (molecular biologists) and low for others (people from IT). Since Transcriptic basically simply charges for reagents on the price list, the main cost difference is labor. The robot already costs quite cheaply per hour, and he is not averse to getting up in the middle of the night to photograph the plate. Once the protocols have been approved, it’s hard to imagine that even a graduate student will be cheaper, especially considering the opportunity costs.
, . , - , . , : , , IDT .
:
, . , :
- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- Expressiveness . You can use the programming syntax to encode repeating steps or branching logic. For example, if you want to dose from 1 to 96 μl of reagent and (96-x) μl of water into a 96-tube plate, this can be summarized.
- Machine readable data . Data with the results is almost always returned in csv or other format suitable for machine processing.
- Abstraction . Ideally, you can run the entire protocol regardless of the reagents used or the cloning style and replace something if necessary, if it works better.
Of course, there are some drawbacks, especially since the tools have just begun to develop. If we compare with the Internet, then we are in the area of 1994:
- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- For many laboratories, it may be more expensive to use a cloud lab than just taking a graduate student to do work (marginal cost per hour: ~ $ 0). It depends on whether the lab needs a graduate student's hands or his intellect.
- Transcriptic is not experimenting at the weekend yet. They can be understood, but this is inconvenient, even if you have a small project.
Protein Making Software
Although there is quite a lot of code and quite a lot of debugging, I think it is possible to create some kind of software that accepts a sequence of proteins at the input, and creates bacteria at the output with the expression of this protein.
For this to work, several things must happen:
- True Twist / IDT / Gen9 integration with Transcriptic (probably, it will be slow due to low demand at present).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
In many applications, you also want to purify your protein (through a column ) or, perhaps, simply force bacteria to secrete it. Suppose that soon we can do this in a cloud lab, or that we can conduct in vivo experiments (i.e., inside a bacterial cell).
There are many possibilities for the protocol to actually work better than a human, for example: the design of promoters and RBS to optimize the expression specific to your sequence; experiment success probability statistics based on comparable experiments; automated gel analysis.
Why all this?
After all this, it may not be entirely clear why create such a protein. Here are some ideas:
- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- Make a local vaccine that enters the body through hair follicles (I do not recommend trying this at home).
- Mutagenicize your protein in hundreds of different ways and see what happens. Then scale up to 1000 or 10,000 mutations? Can characterize GFP mutations?
For new ideas on what's possible when designing proteins, look at hundreds of iGEM projects .
In the end, I would like to thank Ben Miles of Transcriptic for helping to complete this project.
Source: https://habr.com/ru/post/451124/
All Articles