Chemists test retrosynthetic paths fully predicted by AI algorithms.
For the first time, as far as I know, when a computer program predicts synthesis and you go to the lab - bang! - and it works- this is how Bartosh Grzybowski describes his program Chematica.
Prehistory
The idea of computer planning of chemical syntheses is far from new. Elias J. Cory of Harvard University developed the first version of such a program (Logic and Heuristics Applied to Synthetic Analysis) in the 1970s, but it never justified the hopes placed on it. Chematica is one of several new rival software products that have emerged over the past couple of years. Bartosz Grzybowski (Bartosz Grzybowski, Ulsan National Institute of Science and Technology, South Korea and the Polish Academy of Sciences) worked on the program for 15 years before selling it to MilliporeSigma in May 2017.
Usually, chemists rely entirely on their experience and knowledge — chemical intuition — to develop a sequence of reactions that should lead to a large molecule of smaller molecule blocks. Also, chemists must take into account various limitations, such as incompatibility of functional groups in certain reaction conditions.

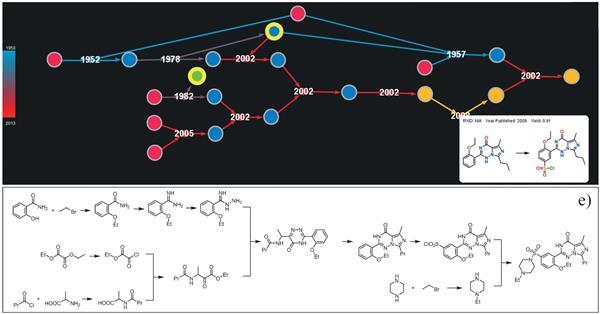
Chematica successfully predicted ways to synthesize eight synthetically complex molecules and in some cases was able to improve the yields of the reaction products.
')
To plan retrosynthesis, according to Grzybowski, is like playing chess: there are a number of basic moves. During the game, each turn opens a new branch to get a different result. But in organic synthesis, “the number of basic steps — the main types of reactions — is simply enormous.” After each synthetic move, there is a choice of about a hundred subsequent possible steps. This means that the more stages of synthesis, the more impressive is the set of possible steps.
But chemists are biased, explains Sarah Trice (Sarah Trice, head of the commercial development technology of cheminformatics from MilliporeSigma - the company recently bought Chematica), that is, there is a tendency to use what has been successful in the past. Grzybowski believes that Chematica will eliminate bias. The Chematica algorithm has been trained by more than 50,000 rules over the past 15 years, as well as a means of finding options in this vast chemical space and developing the right reaction sequences.
Tests
Despite the attractiveness of the concept, there was no data to confirm that it really works. But now Chematica has proven significant in laboratory tests. The algorithm found important paths for eight molecules: six small bioactive compounds, one blockbuster drug, and one natural compound molecule.
The early suggested paths for most of these six medicinal compounds had problems with low yield of products, and some of them were not synthesized at all. Chematica has developed paths with less than 10 synthetic steps, using only conventional reagents. However, the team was able to get targeted products with high yields - in some cases from 1% to 60% - while saving time and money for laboratory tests, compared with previous attempts.
Some of the retrosynthetic decompositions of the program were unusual — such as the three-component aza- henry reaction in one of the paths to a single quinolone-articulated lactam stereoisomer. “It was unusual for chemists to carry out some stages of the synthesis, as their instinct told them that this might not work out,” Tris laughs. Grzybowski says that "the rules of the game were as follows: you cannot change retrosynthetic divisions and you need to follow a common methodology."
Restrictions
Since Chematica does not provide the exact conditions for each reaction, everything is necessary to go through trial and error when it comes to optimizing the synthesis. However, for realism, time and finances were limited to five attempts for each reaction and a maximum of 70 hours for carrying out each complete synthesis.
The blockbuster drug against arrhythmia drug dronedarone (dronedarone) served as an example of another problem: its synthesis is protected by 46 patents.

Chematica was able to plan the synthesis of the drug dronedarone against antiarrhythmias, which avoided all variants protected by patents. The program also knows how to take into account the cost of reagents for production and choose cheaper ways of synthesis.
Richmond Sarpong (University of California, Berkeley) and his research team also got a chance to test a new instrument. “We were very impressed with the possibilities. The algorithm is especially effective in finding solutions to certain structural fragments. In this part, I find that the program is more capable than a person. ”
Nevertheless, Sarpong points out that “like people, the program tries to predict with the participation of architecturally complex molecules where stereo-electronic subtleties arise”. The Sarpong team is currently working on one of the synthetic pathways predicted by Chematica.
Grzybowski believes that the next step will be complex natural compounds. His team completes the 15-step synthesis of the newly isolated natural alkaloid. “I never considered myself a very competent organic chemist. This is the first complete synthesis in my life, ”he says. “I believe that this can really help people who have not received the classical training of a synthetic chemist,” adds Tris.
Grzybowski readily notes that Chematica should not be considered a threat to the experience of chemists. Since the algorithm cannot teach itself new reactions, “we still need courageous organic chemists who are ready to challenge such programs as Chematica and find unconventional ways to create molecules,” agrees Mariola Tortosa (organic chemist specializing in the synthesis of natural compounds at the university Madrid, Spain). “However, the program can significantly speed up [retro-synthesis],” she says.
John Maxwell, vice president of chemistry at Tango Therapeutics, believes that this article does not prove that Chematica is planning better syntheses than chemists. He points out that the chemists whose synthesis was chosen for comparison did not necessarily optimize the synthesis for product yield or the length of the reaction chain.
Some chemists are wondering how Chematica and MilliporeSigma will address intellectual property issues. Richmond Sarpong says that researchers may not dare to use Chematica, since it is not clear what company will have access to molecules that users enter or who will own the intellectual property of the synthetic pathways that Chematica generates. Sarah Tris explains that only the user can see the target molecules, and that MilliporeSigma will not control the intellectual property of the synthesis options offered by Chematica.
The company is already working with industry and academic software testing partners and hopes to release a commercial version later this year.
The original article is in the public domain. 10.1016 / j.chempr.2018.02.002
To whom it is interesting, here you can read how retrosynthesis is planned.
Video example of the program interface
Criticism, corrections and suggestions are welcome.
Error is better to write in private messages, so as not to distract people who are looking for additional information or feedback in the comments. The translation is not literal. Some paragraphs are abbreviated.
- Added a paragraph from the news story https://cen.acs.org/articles/96/i10/Chemists-test-computer-planned-syntheses.html .
- Added two more paragraphs with comments at the end of the post.
- Added video.
Source: https://habr.com/ru/post/410781/
All Articles