Genetic engineering of bacteria: how to get the protein we need from bacteria
So, in the two previous articles on the genetic engineering of bacteria ( one and two ), we figured out how to collect the genes we need, in what form they can be introduced into a bacterium, and how exactly they can be inserted there. Suppose we did all these manipulations in order to make a biofactory for the production of protein. Then now it’s easy to get the bacteria out of our bacteria in the purest form possible.
There are many methods for solving this problem, most of them are related to chromatography. How these methods work read under a cat.
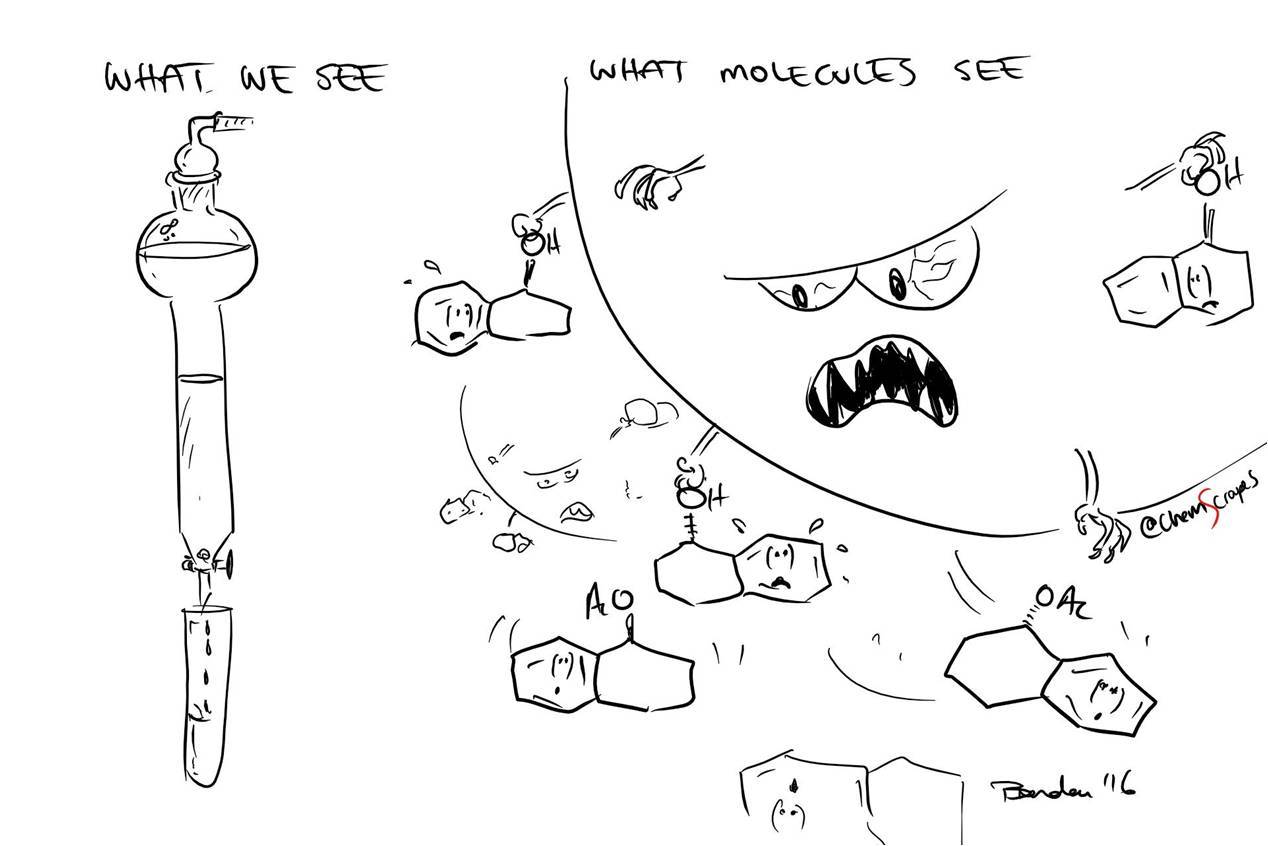
The stationary phase "catches" molecules passing by for their (-OH) groups.
So, we got the strain we needed and increased the biomass. Now we need to somehow remove our protein from it.
First of all, we take our culture and pour it into bottles for centrifugation. Bacteria are rather large and heavy objects, so for their precipitation no monstrous overload values are necessary when centrifuging: 10,000g will be enough for more than 20 minutes. As a result, we obtain a precipitate at the bottom of the vials and liquid (it is called the supernatant). We pour out the liquid, because our protein is in the bacteria that are in the sediment. Then you can act on the circumstances: either immediately work with the obtained sediment, or freeze and store until it is needed. It can be stored for a very long time. The further procedure of working with sediment does not depend on whether we kept it in the freezer or not.
Let's say it's time to work with sediment. What's next?
')
First of all, we need to destroy the cells so that the protein can get out. To do this, use the following methods:
From my own experience I can say that sonication in combination with detergents is usually more than enough.
So, we got a cell lysate - “soup”, consisting of the protein we need and a huge amount of “garbage” (chromosomal and plasmid DNA, pieces of the cell wall, various proteins, etc.). How to get rid of all this?
The first question that needs to be answered is: “In what form did our protein exist in the cell?”. There are two main options:
So, we have ready some volumes of protein solutions in the folded state. The further procedure is the same for any "initial conditions" - we filter our solutions through filters with a pore size of 100-450 nm (in order not to clog the chromatographic columns and the chromatograph itself), and then perform chromatography.
The principle of any chromatography is that the various components of the solution move through the column at different speeds; as a result, the components “entering the column” together end up at the output at different times.
But first, we recall another method - protein electrophoresis in a gel.
This method allows the separation of proteins according to their masses. For this purpose, a buffer containing SDS and a dye (usually bromophenol blue) is added to the sample. At the same time, the density of the buffer is higher than the density of water; this is done to make the mixture of the sample and buffer easier to apply to the wells on the gel. The dye is added to control the procedure for introducing the sample into the well, and it also serves as an indicator that it is time to finish the Forez: the ink moves faster than the protein, so when it reaches the end of the gel, the Forex stops. This is very convenient, since we do not see the movement of the protein along the gel.
SDS-PAG electrophoresis. It is better to read and see once than read a hundred times. The molecular weight marker was applied to the first well on the forez in the video, and the samples under study were applied to the remaining wells.
SDS is a key component: this negatively charged detergent (surfactant) denatures proteins, after which it “surrounds” them, and it turns out that the number of SDS molecules attached to a protein is directly proportional to its mass. Then it follows from Coulomb's law that in the absence of any resistance to movement, all proteins surrounded by SDS molecules would have the same acceleration in an electric field (the force grows linearly with the charge, but linearly with the charge the mass grows).
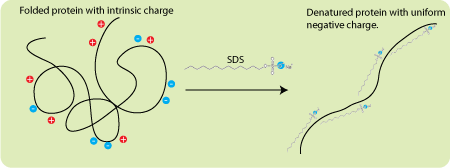
Diagram of the effect of negative charged detergent SDS on protein. First, he denatures him, and then “envelops” him. As a result, the protein consists of a negatively charged membrane consisting of SDS, the charge of which is directly proportional to the mass of the protein.
And here we come to the aid of PAGE (PAG - polyacrylamide gel). During polymerization, it forms a network of channels of approximately the same diameter, through which, all other things being equal, small objects move faster than large ones. It is obvious that the heavy protein is larger than the lung, so light proteins move faster.
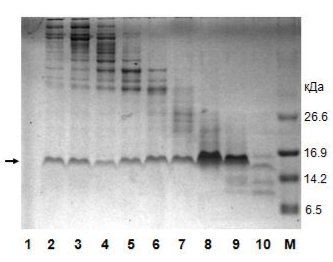
After the end of the electrophoresis procedure, the gel is stained. More precisely, the places where the protein is in the gel are stained - blue stripes are obtained (or black, depending on what to paint). In order to understand what mass corresponds to one or another strip, you need to compare with something, therefore, so-called is applied to each gel. "Molecular weight marker" - a mixture of components with known masses.
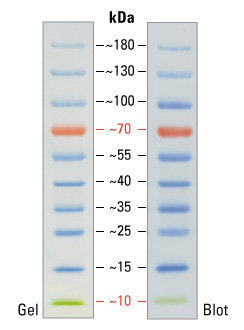
An example of molecular weight marker strips. This marker is also convenient due to the fact that its two bands are painted in an individual color: red (70 kilodaltons) and green (10 kilodaltons). This greatly helps the experimenter to navigate the marker itself. 1 dalton, also known as atomic mass unit, is defined as 1–12 mass of a free carbon 12 atom at rest, which is in the ground state.
It would seem a miracle, not a method - the strips do not overlap, which means that the proteins are ideally divided! But in fact, scientists have not learned how to conduct protein electrophoresis on an industrial scale, and therefore we still use column chromatography.
An approximate scheme of any chromatography. A column is something filled with a cylinder. This “something” is called a “stationary phase,” and the “mobile phase” is a solution, the components of which we need to separate. Components move at different speeds and therefore leave the column separately.
Most chromatographic techniques are based on the fact that passing by proteins cling to the fixed filler of the column "with different enthusiasm": those who "cling" often and strongly move the slowest of all, and those who do not "cling" to anything that quickly leave the column. As a result, a substantial part of the mixture often leaves the column without ever “catching on” for it - they go together at the very beginning of chromatography (this fraction is called “overshoot”, as they slipped through the column without “catching on” for it). But what to do with those who "hooked"?
To begin with, let us deal with what is happening in the column with those who “cling”. The “hook” itself occurs because there is something on the surface of the stationary phase that is capable of “capturing” squirrels passing by for a while (how it happens - just below). That is, the “clinging” protein spends some time in a state associated with a solid phase, then it detaches from it (due to its own thermal motion, collisions, etc.), swims further, again binds to the solid phase, again detaches and so on until he leaves the column. It is clear that the more often and the stronger it is associated with the stationary phase, the slower it moves along the column.
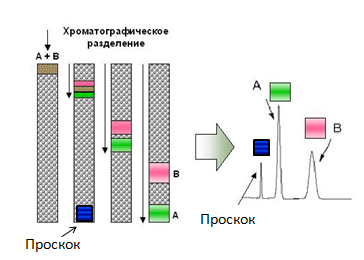
The principle of chromatography. That which does not bind to the column (overshoot) moves at the speed of the mobile phase (adjusted for size). The binding components have different speeds, which leads to their separation at the output.
Generally speaking, only in the case of great bad luck there will be different components in the mixture that move with the same speed, so we can just wait for them to come out of the column. But it is often “just to wait” for a very long time, as the components move slowly. In this case, they need to be “customized”, and the way they are “customized” depends on the type of the column.
Finally, consider the chromatographic methods themselves, which are suitable for the industrial production of belov.
Well then. , , .
There are many methods for solving this problem, most of them are related to chromatography. How these methods work read under a cat.
The stationary phase "catches" molecules passing by for their (-OH) groups.
So, we got the strain we needed and increased the biomass. Now we need to somehow remove our protein from it.
First of all, we take our culture and pour it into bottles for centrifugation. Bacteria are rather large and heavy objects, so for their precipitation no monstrous overload values are necessary when centrifuging: 10,000g will be enough for more than 20 minutes. As a result, we obtain a precipitate at the bottom of the vials and liquid (it is called the supernatant). We pour out the liquid, because our protein is in the bacteria that are in the sediment. Then you can act on the circumstances: either immediately work with the obtained sediment, or freeze and store until it is needed. It can be stored for a very long time. The further procedure of working with sediment does not depend on whether we kept it in the freezer or not.
Let's say it's time to work with sediment. What's next?
')
First of all, we need to destroy the cells so that the protein can get out. To do this, use the following methods:
- Sequential freeze-defrost. Not the most effective way to destroy the cell wall, but it is safe for protein, so they often start with several such cycles and then move on to more efficient methods;
- Ultrasound treatment (sonication). The method is widely used, it is simple and effective. Usually, ultrasound is fed directly into the volume of water using an elongated "speaker", which is inserted into the fluid with the cells so that the end of the "speaker" almost reaches to the bottom. This is a rather “hard” method. There is also a "softer" alternative - ultrasound is fed into a water bath in which the tubes float.

MIT graduate warns - watch your sonicists carefully.
When carrying out ultrasonic destruction of cells, it is important to monitor the temperature: water heats up quickly under the influence of ultrasound, and we do not need denaturation of our protein at all, and all the more we would not want to boil it. Therefore, in the first case (with an “elongated column”), a tube with cells is inserted into a cup with ice and water, and there should be plenty of ice. Water in this case acts as a thermal interface between the ice and the tube cooled by it. In the case of the “soft alternative”, ice is added directly to the water bath.
Demonstration of "hard" ultrasonic processing. Pay attention to the creepy noise standing around the installation, it is especially well heard at 2:06 of this video. Someone who can save himself is trying to: someone tries to go out of the room for the time of sonication, someone wears special noise-reducing headphones.
Demonstration of "soft" ultrasonic treatment. - Adding lysing agents to the medium. These are mainly detergents, that is, “soap” (in general, the word “detergent” in English means “detergent”). Detergents are essentially related to lipids in cell membranes. Once in the medium with the cells, the detergents begin to be embedded in the membrane, which leads to their destruction. And we just need this. Sometimes lysozyme is also added - an enzyme that destroys the cell wall of bacteria.
- French press. The cell suspension is sucked through the inlet valve into the working cylinder, from where it is squeezed by a press through a narrow slit. The transverse forces that occur when pressure drops to atmospheric pressure when cells pass through the gap, lead to the destruction of the cell wall and membrane.
From my own experience I can say that sonication in combination with detergents is usually more than enough.
So, we got a cell lysate - “soup”, consisting of the protein we need and a huge amount of “garbage” (chromosomal and plasmid DNA, pieces of the cell wall, various proteins, etc.). How to get rid of all this?
The first question that needs to be answered is: “In what form did our protein exist in the cell?”. There are two main options:
- The protein was soluble before lysis and with a very high degree of probability remained so after it, which means that we cannot sediment it by centrifugation. But on the other hand, we can precipitate large “trash” remaining after lysis: this way we will get rid of pieces of the cell wall, chromosomal DNA and other large fragments. The precipitate is discarded, and the supernatant is sent to chromatography;
- Protein in the conditions of bacterial cytoplasm was insoluble and it formed the so-called. "Inclusion bodies", preserved after lysis. Inclusion bodies are protein balls that are formed if the protein does not have time to fold (that is, to take its normal soluble form) during the synthesis, before it collides with another similar unfolded protein. Non-folded proteins almost always have hydrophobic regions sticking out, with which they will stick to each other. After the two squirrels have "stuck together", they no longer have enough degrees of freedom to complete their folding. Then the third, fourth, fifth ... swim to them. The result is a protein granule. This usually happens when we synthesize eukaryotic proteins in bacteria (as I wrote earlier, the bacterium does not know how to produce them). As a result, we have a mass of protein with a “poor” spatial structure. But there is a blessing in disguise, because as a result, granules are formed that consist almost entirely of the protein we need!
Since, in this case, our protein is insoluble, it is in the sediment when centrifuged along with the “garbage”. We discard the supernatant, and then the bodies in the sediment are washed off from all unnecessary things as follows:- Pouring wash buffer No. 1 was poured in which the “garbage component of sediment No. 1” is dissolved;
- Stir the precipitate until homogeneous and set on a shaker (a device that shakes);
- Centrifuged and poured the supernatant. In the end, what was dissolved in the wash buffer No. 1 was separated from the sediment;
- Pouring wash buffer No. 2 was poured in which the “garbage component of sediment No. 2” is dissolved;
- Well, and so on. In total, about 5 washes are usually needed.
Let me remind you once again that if we are dealing with inclusion bodies, then we are dealing with an unfolded protein, which means that it is not able to fulfill its functions. Therefore, before subsequent purification, it is refolded, that is, it is brought into active form. The most common refolding method consists of the following steps:- Dissolve the calf in "non-physiological" conditions. Usually, the calves dissolve well at high pH values (about 12) in the presence of two-molar urea. The idea of this method is that at high pH there are very few protons in the medium and everyone who could get rid of them can easily do it. A proton is positively charged, which means that all proton donors receive a negative charge, and then everything that makes Coulomb's law does — proteins that have a large negative charge start to repel each other;
- Drop by drop add the entire volume of the “protein solution” from the first item to a much larger actively mixed volume of buffer with protein-comfortable conditions. The idea of this stage is that at a low protein concentration, he is much more likely to fold before he meets with his unfolded counterpart. Usually, the drop-by-step transfer of the protein solution is carried out in buffer volumes exceeding the volume of the solution itself at least 10 times.
So, we have ready some volumes of protein solutions in the folded state. The further procedure is the same for any "initial conditions" - we filter our solutions through filters with a pore size of 100-450 nm (in order not to clog the chromatographic columns and the chromatograph itself), and then perform chromatography.
Types of chromatography
The principle of any chromatography is that the various components of the solution move through the column at different speeds; as a result, the components “entering the column” together end up at the output at different times.
But first, we recall another method - protein electrophoresis in a gel.
This method allows the separation of proteins according to their masses. For this purpose, a buffer containing SDS and a dye (usually bromophenol blue) is added to the sample. At the same time, the density of the buffer is higher than the density of water; this is done to make the mixture of the sample and buffer easier to apply to the wells on the gel. The dye is added to control the procedure for introducing the sample into the well, and it also serves as an indicator that it is time to finish the Forez: the ink moves faster than the protein, so when it reaches the end of the gel, the Forex stops. This is very convenient, since we do not see the movement of the protein along the gel.
SDS-PAG electrophoresis. It is better to read and see once than read a hundred times. The molecular weight marker was applied to the first well on the forez in the video, and the samples under study were applied to the remaining wells.
SDS is a key component: this negatively charged detergent (surfactant) denatures proteins, after which it “surrounds” them, and it turns out that the number of SDS molecules attached to a protein is directly proportional to its mass. Then it follows from Coulomb's law that in the absence of any resistance to movement, all proteins surrounded by SDS molecules would have the same acceleration in an electric field (the force grows linearly with the charge, but linearly with the charge the mass grows).
Diagram of the effect of negative charged detergent SDS on protein. First, he denatures him, and then “envelops” him. As a result, the protein consists of a negatively charged membrane consisting of SDS, the charge of which is directly proportional to the mass of the protein.
And here we come to the aid of PAGE (PAG - polyacrylamide gel). During polymerization, it forms a network of channels of approximately the same diameter, through which, all other things being equal, small objects move faster than large ones. It is obvious that the heavy protein is larger than the lung, so light proteins move faster.
After the end of the electrophoresis procedure, the gel is stained. More precisely, the places where the protein is in the gel are stained - blue stripes are obtained (or black, depending on what to paint). In order to understand what mass corresponds to one or another strip, you need to compare with something, therefore, so-called is applied to each gel. "Molecular weight marker" - a mixture of components with known masses.
An example of molecular weight marker strips. This marker is also convenient due to the fact that its two bands are painted in an individual color: red (70 kilodaltons) and green (10 kilodaltons). This greatly helps the experimenter to navigate the marker itself. 1 dalton, also known as atomic mass unit, is defined as 1–12 mass of a free carbon 12 atom at rest, which is in the ground state.
It would seem a miracle, not a method - the strips do not overlap, which means that the proteins are ideally divided! But in fact, scientists have not learned how to conduct protein electrophoresis on an industrial scale, and therefore we still use column chromatography.
An approximate scheme of any chromatography. A column is something filled with a cylinder. This “something” is called a “stationary phase,” and the “mobile phase” is a solution, the components of which we need to separate. Components move at different speeds and therefore leave the column separately.
Most chromatographic techniques are based on the fact that passing by proteins cling to the fixed filler of the column "with different enthusiasm": those who "cling" often and strongly move the slowest of all, and those who do not "cling" to anything that quickly leave the column. As a result, a substantial part of the mixture often leaves the column without ever “catching on” for it - they go together at the very beginning of chromatography (this fraction is called “overshoot”, as they slipped through the column without “catching on” for it). But what to do with those who "hooked"?
To begin with, let us deal with what is happening in the column with those who “cling”. The “hook” itself occurs because there is something on the surface of the stationary phase that is capable of “capturing” squirrels passing by for a while (how it happens - just below). That is, the “clinging” protein spends some time in a state associated with a solid phase, then it detaches from it (due to its own thermal motion, collisions, etc.), swims further, again binds to the solid phase, again detaches and so on until he leaves the column. It is clear that the more often and the stronger it is associated with the stationary phase, the slower it moves along the column.
The principle of chromatography. That which does not bind to the column (overshoot) moves at the speed of the mobile phase (adjusted for size). The binding components have different speeds, which leads to their separation at the output.
Generally speaking, only in the case of great bad luck there will be different components in the mixture that move with the same speed, so we can just wait for them to come out of the column. But it is often “just to wait” for a very long time, as the components move slowly. In this case, they need to be “customized”, and the way they are “customized” depends on the type of the column.
Finally, consider the chromatographic methods themselves, which are suitable for the industrial production of belov.
- Ion exchange chromatography. The idea of the method is that the stationary phase has a charge under the conditions of chromatography. If the charge is positive, then they say about "anion-exchange chromatography", if negative, then about "cation-exchange".
The logic of the name is simple, consider it on the example of an anion exchanger: negatively charged components of the mixture “stick” to the positively charged solid phase of the column. Moreover, if we follow a specific positively charged group on the surface of a solid phase, then one or another negatively charged component will “stick” to it one by one. Thus, there is a kind of anion exchange between the stationary and mobile phases, hence the name “anion-exchange chromatography”.
Obviously, if we want to use anion-exchange chromatography, then we must carry out the separation of the mixture in such conditions under which the solid phase is positively charged. At the same time, we have a choice of two options for further development of events depending on what kind of charge a protein has under these conditions: if “-”, then it “sits” on the column, and if “+”, then it will be in overshoot.
How to speed up the output of related components, that is, how to carry out the "elution"? You can act in two ways:- You can change the charge of the stationary phase and the bound components by changing the pH of the mobile phase. It is important to make the process of pH change gradually, then the components will accelerate their movement, but they will not “fall out” at the same time all at once;
- Add other anions to the column. Indeed, the column has a finite number of places for which anions can catch, and if we take all these places with something, the components of the mixture will move faster. For anion exchange chromatography, the addition of chlorine anions is typical. The addition of competing anions should also be carried out gradually.
The procedure for cation exchange chromatography is generally the same, only the opposite is true.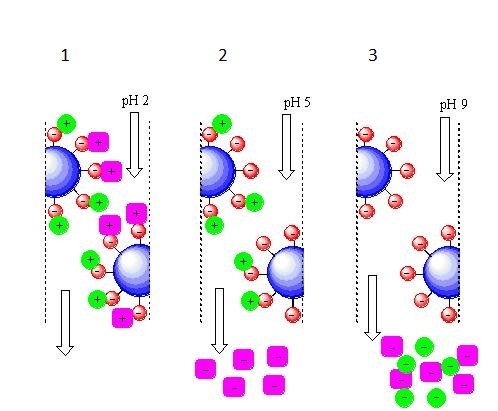
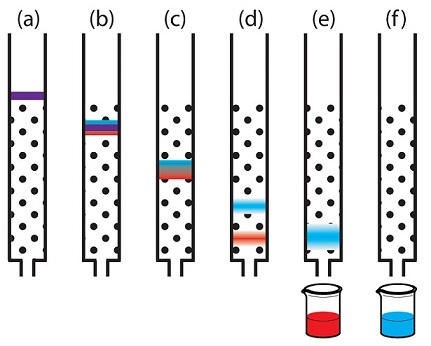
Cation-exchange chromatography with elution due to changes in the pH of the mobile phase.
1 - at pH = 2, there are a lot of protons in the mobile phase, so all the components of the mixture picked them up from the liquid, while receiving a positive charge;
2 - at pH = 5, the number of protons in the mobile phase has decreased and the “purple” component already acts not as an acceptor, but as a proton donor, therefore, it has a negative charge at a given pH value and it leaves the column. The “green” component still acts as a proton acceptor and it retains a positive charge at a given pH value, which means it is still holding on to the column;
3 - at pH = 9, there are very few protons in the mobile phase, the “green” component also becomes a proton donor, acquires a negative charge and leaves the column last.
In general, the choice of the type of ion exchanger and the conditions of separation depends on what we want to isolate and what else is in our mix. - You can change the charge of the stationary phase and the bound components by changing the pH of the mobile phase. It is important to make the process of pH change gradually, then the components will accelerate their movement, but they will not “fall out” at the same time all at once;
- Another separation method is based on the hydrophobicity (~ non-polarity) and hydrophilicity (~ polarity) of certain substances. The idea here is exactly the same as in the case of the ion exchanger: the components, according to their physical properties, are “torn” between hydrophobic and hydrophilic environments, some linger longer, some less.
First of all, let me remind you that the non-polar dissolves in the non-polar, and the polar in the polar. Often, “hydrophobic” and “non-polar” are the same as “hydrophilic” and “polar”, although in general this is not the case. An example of a non-polar solvent is ethanol, and an example of a polar one is water.
The polar bear laments the fact that it dissolves in water, which is a polar solvent. "Non-polar" brown bear is not threatened.
Now consider an example. Suppose the stationary phase is hydrophobic (non-polar). Then the mobile should be generally hydrophilic (polar). All hydrophobic components will “sit down” on the column, since it is “scary” to swim in the hydrophilic mobile phase. Hydrophilic components will be in the overshoot, as they prefer the hydrophilic mobile phase. Then, the hydrophobic components “sat down” on the column should be eluted by gradually increasing the hydrophobicity of the mobile phase. The regulation of the hydrophobicity of the mobile phase is carried out by a pump that simultaneously pumps the polar and non-polar solvent from two different tanks through the column. The total polarity of the mobile phase depends on the ratio between them. - Metal chelate chromatography with his-tag (His-tag). The method is based on the fact that areas of six histidines (histidine is one of 20 proteinogenic amino acids) bind well to the nickel ions on the column through their imidazole groups. The method is convenient because there is little in which natural protein there is a histidine tag, which means with a high degree of probability all proteins except ours will be in the breakthrough.
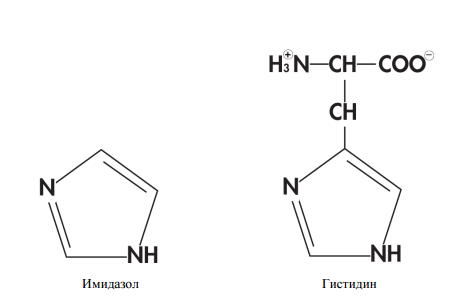
The structure of the molecules of imidazole and histidine.
The disadvantage of the method is that it is necessary to modify the protein, and this may affect its properties. Addition of 6 histidine tags to the protein occurs at the stage of gene creation, and to minimize the effect of the tag on the protein, these amino acids are added at the beginning or end of the amino acid sequence. Naturally, the method will not work if the protein is such that its beginning and end are hidden inside: the nickel ions and the tag simply cannot meet.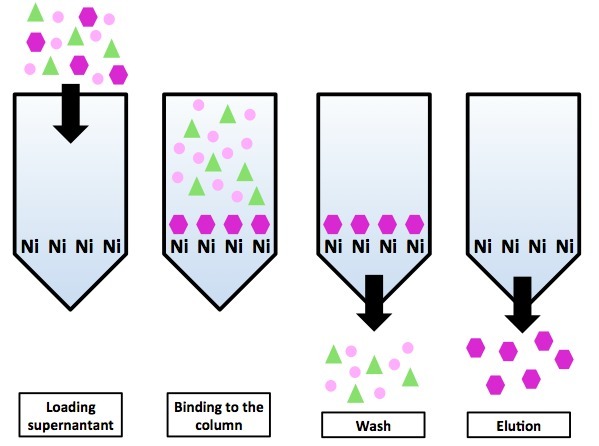
Chromatography on a nickel column using his-tag tag. All proteins, except for the “purple hexagon” protein, do not have His-tag and are in the gap. The purple hexagonal proteins are eluted by imidazole coating on the column.
"Remove" protein from the column can be a gradual addition of imidazole. The essence of the elution method is the same as that of ion exchangers - imidazole competes with the His-tag protein for binding sites on the stationary phase (only on the ion exchanger binding sites were abstract ions, and on the metal chelate column, the binding sites are nickel ions). As a result, it occupies most of the available binding sites, and the squirrel has no choice but to leave the column.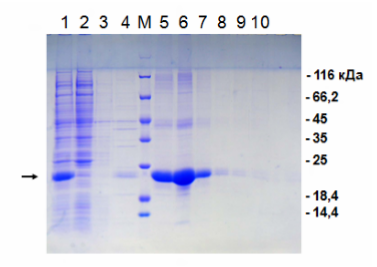
Electrophoresis according to the results of nickel metal chelate chromatography.
1 - the original "dirty" material for application to the column; 2 - overshoot; 3 and 4 - washing without imidazole; M - molecular weight marker; 5-10 sequential elution with imidazole. The protein we need is labeled with an arrow to the left. As we see, in the breakthrough there is no our protein, but in it there is a lot of "garbage". The eluate is a purified target protein. On the right are the masses of the respective bands of the molecular weight marker.
The method could potentially be convenient also by the fact that there is a dependence of the binding force on the amount of histidine tags in the protein composition. Thus, it would be possible to attach to one protein tag only at the beginning, and to the other from both ends and synthesize them in one bacterium. But in reality it is not very practical: the difference in the concentrations of imidazole that elute such proteins is small (approximately 75 mM and 100 mM imidazole for one and two tags, respectively), so there is a possibility of difficulties with their separation from one mixture. And making two separate strains is no more difficult than making one with two genes.
Another possible difficulty in working with the His-tag is the need to obtain a protein without any modifications, which exists in some experiments. In principle, it is possible to cut off the protein from the enzymes, but some of the protein will remain with the tag (that is, “EFFICIENCY <1”), and this will also lead to the fact that we will need to carry out an additional stage of purification from these enzymes. In addition, each stage of purification is associated with losses of the target product. - Gel filtration I’ll just say that this method can be called suitable for protein extraction only conditionally: if we need to get a small amount of protein for research, then using gel filtration makes sense, but this technique is not suitable for working with truly industrial volumes, and here's why.
In all previous methods, the speed of movement of the components associated with a column in the absence of “fitting factors” is so small that it can generally be considered close to zero. With gel filtration, everything is different: all components, even the largest ones, move at quite noticeable speeds. , - - , ( , ), . : - 1-2% . , , , 10 400 : , . - .
-: , .
- ? , , . , , , . - , .
-. . , . — . .
-, . , , , . - — , .
Well then. , , .
Source: https://habr.com/ru/post/402685/
All Articles