Algorithm for the selection of traits for the DNA test
In scientific research, as in any other kind of meaningful human occupation, a plan of action is important. There are people who are engaged in pure science, but this is a completely different story. When it comes to the application, that is, the use of the scientific method in everyday life, we need a clear sequence of actions, in accordance with which the pipeline will work. Genotek is engaged in applied genetic research, analyzing and interpreting data recorded in DNA. Our scientific department has published an article about how to choose the right genes and why the results of the analysis of these genes can be trusted. Here is a brief summary of the publication.

Sometimes surprises (and even troubles) occur in our lives, but if we treat our actions responsibly, then we can recognize that a significant part of what is happening is a result of our own actions, and not a chain of random events. In general, this is a matter of confidence. You can never worry, do not doubt, do not think about these things and do something beautiful. On the other hand, the ability to find your own instructions for efficient operation opens up new possibilities. It's very simple: there are a few things that a person wants to know for sure.
All this can be learned from DNA by reading certain parts of the genes. A gene is a separate sense DNA fragment, which in the simplest case encodes a single trait. By the word "phenotype", by the way, we should understand the totality of all the signs of the observed state (for example, eye color, shape of the lobe, tendency to diabetes, lactose digestion). And then the same question arises as in school years: what exactly needs to be read, is it possible to read not everything and how to deal with what has been read.
')
To make a mass medical service out of the technology of prognostic DNA tests, you need to pay attention to a few subtle points in data analysis. First, this is the problem of personalization: it is necessary to select markers carefully. Most studies aimed at establishing the genotype-phenotype association are conducted on large samples. Samples (analyzed groups of people), as a rule, are not very heterogeneous : they can be voluminous, but consist of representatives of one ethnic group. On the one hand, this simplifies the statistical analysis within the framework of the study, on the other hand, it raises the question of the relevance of the detected association in relation to other groups of people.
The second problem is the number of markers - the very case when you need to find a middle ground. And here we can not do without the eternal balancing between the two test parameters: sensitivity and specificity. An increase in the number of genetic markers undoubtedly increases the sensitivity of the assay to certain diseases. And at the same time, the specificity of the test may fall. Since we are talking about, for example, identifying a predisposition to serious diseases, a false positive error in identifying a terrible diagnosis will be more dangerous than specifying the likelihood of developing a disease. In addition, an increase in the number of markers leads to an increase in the cost of the test system, which also complicates access to the mass market.
There is no universal solution for these problems. At the initial stage of work, the researcher is faced with a situation where “there are a lot of articles, and they all need to be read.” For this reason, Genotek’s science department has proposed a selection algorithm that greatly simplifies the selection of polymorphisms for trait analysis. It is important that we are talking about polymorphisms, not about the genes entirely: two men have the same androgen receptor gene on each of the X chromosomes, but the first man has adenines (AA) in the rs6152 locus and the guanines in the second (GG). The likelihood that the last head of hair by 40 years will remain as lush is about 30% . In this case, to understand this, you do not need to read the entire gene sequence - just find and read only one point on the DNA, and then compare it with the one opposite.
Geneticists call phenotypic peculiarities that we adopt from our ancestors. The fundamental problem of biology as a whole is the relationship between genotype and phenotype, as well as how one encodes the other. In our case, there are two views on the heritability of phenotypic features. On the one hand, the nature of inheritance may differ. Thus, a single trait may not be inherited at all, and its manifestation will in no way be associated with a genetic contribution. Such signs arising under the influence of lifestyle or external environmental conditions are of no interest to the developers of DNA tests.
On the other hand, if the trait is inherited, then it is important to understand what kind of genetic interaction underlies the observed state. In the simplest case, only one gene can influence the development of the disease - then we are talking about monogenic inheritance. For example, phenylketonuria is a metabolic disease that develops due to a “breakdown” in one gene. In this case, you can not talk about predisposition: if a person already has two copies of a broken gene, he will still develop the disease. However, in such a situation, genetic diagnosis can clarify the diagnosis for choosing a more adequate treatment.
It is more difficult when several genes contribute to the phenotypic feature. It is important to understand how the formation of the trait takes place, whether it is polygenic or multifactorial. Polygenic trait develops as a result of several polymorphisms - there is also no probabilistic nature of the development of the trait. With a certain combination of polymorphisms, you can reliably get a certain color of human eyes. Now the problem of predicting eye color (and other external features) is in forensic science. It is solved both with the help of mathematical models, and by training neural networks on large data sets. The multifactor phenotype, in addition to the genetic basis, develops under the influence of certain environmental conditions. These signs are of the greatest interest for medical prognostic analysis, since these are conditions that can be corrected with the help of a certain lifestyle.
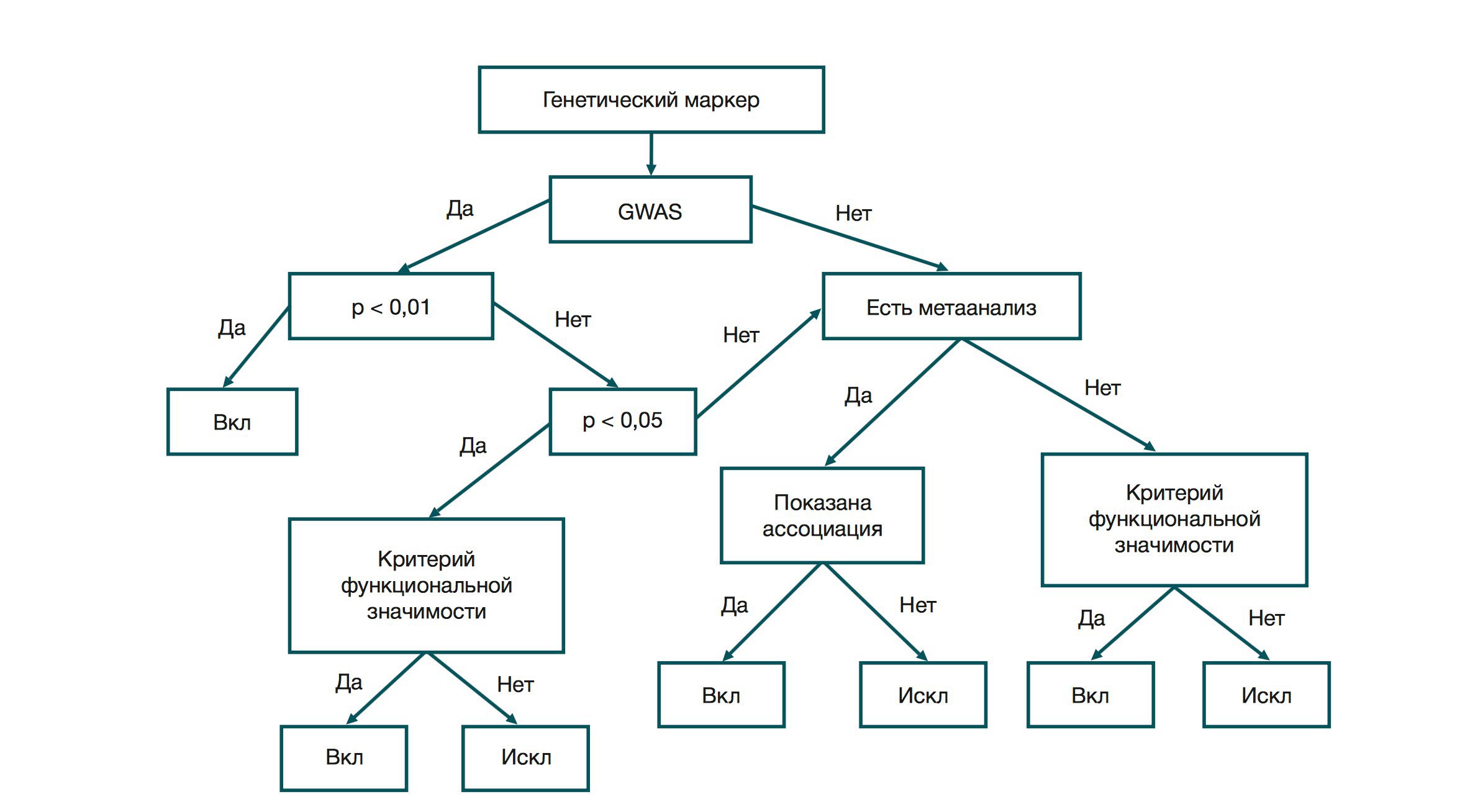
The first stage is a check for the presence of a genome wide association study (GWAS). This line of genetic research has been actively used with the development of microchipping technology. The purpose of the study is to find differences between the genomes of people with and without a specific trait. GWAS studies are characterized by large samples. For example, about 300,000 people took part in the GIANT (Genome Investigation of Antropometric Traits - a genomic study of anthropometric traits). The study confirmed the serious influence of the genetic factor on the development of obesity. The probability of observing the genotype-trait association in patients and healthy subjects, provided the association hypothesis is incorrect, is the p-value of the experiment. If this GWAS study value is less than 0.01 (adjusted for multiplicity), the polymorphism is placed on the short list of disease markers. If it is in the range of 0.01–0.05, compliance with one of the criteria of functional significance is required.
Criterion one: the direct or indirect mechanism of the influence of polymorphism on the trait is known. If the metabolic pathway of a substance is known (that is, the chain of transformations of substance A into substance X), then knowledge of which enzymes are involved in this chain may indicate the relevance of this polymorphism. For example, we know an enzyme that activates folic acid to catalyze the conversion of homocysteine to methionine. Methionine is an essential amino acid, and the accumulation of homocysteine can damage the endothelium of blood vessels. That is, we can talk about the replacement in the locus that encodes the active center of the enzyme, as the reason for the decline in homocysteine metabolism. Moreover, the presence of a single polymorphism of this kind does not guarantee the development of the disease.
If, according to the GWAS results, the value of the p-value of the association is greater than 0.05, the meta-analysis data are required. Meta-analysis is a study in which an association was identified by summarizing the results of a multitude of studies. If the meta-analysis shows the association “genotype-phenotype,” polymorphism is taken into work. If there is no data on meta-analysis, polymorphism is tested again by the criterion of functional significance.
Since the bulk of the work is the study of publications, there are strict criteria for the selection of scientific articles to find associations. For GWAS studies of these criteria 3:
For publications aimed at identifying compliance with the criterion of functional significance, the criteria are as follows:
The results of the meta-analysis, if any, are in priority. The requirements for meta-analysis are also very high: the publication itself must carefully select the sources, among which are both confirming and disproving the association of polymorphism with the attribute. Sources of information and keywords for which the search was conducted should be indicated, and justification of the criteria for inclusion and exclusion of publications (sample size, articles in English only, demographic characteristics of participants, etc.) is required. Special attention is paid to the so-called analysis of the bias of individual publications by the method of a funnel graph or sensitivity analysis. When the marker is included in the analysis, we focus on which populations took part in the study.
Despite the fact that the technology itself has long existed on the market, this is not some kind of “routine practice” according to which tons of manuals are written. The DNA test pipeline is working, and this is one of its sections. We will gradually introduce you to each of its fragments.
Sometimes surprises (and even troubles) occur in our lives, but if we treat our actions responsibly, then we can recognize that a significant part of what is happening is a result of our own actions, and not a chain of random events. In general, this is a matter of confidence. You can never worry, do not doubt, do not think about these things and do something beautiful. On the other hand, the ability to find your own instructions for efficient operation opens up new possibilities. It's very simple: there are a few things that a person wants to know for sure.
- First, for which diseases it is most susceptible. Age-related diseases are a consequence of lifestyle, and not someone else's intent.
- Second: the health of children. Again, it makes sense to talk about it when it comes to a responsible person who consciously plans his family, and does not save on contraception, ignoring the consequences.
- Third: what to do to live better. The human body is a mechanism. And although the principles of work are the same for all, they may differ somewhat from body to body. What suits one person may be harmful to another.
- Fourth: what "I" is. From the point of view of functioning, knowledge of one’s origin does not help to live longer or better. Rather, it will allow you to feel yourself a character in the history of mankind, a hero, acting and developing regardless of someone's short-term interests or personal convictions.
All this can be learned from DNA by reading certain parts of the genes. A gene is a separate sense DNA fragment, which in the simplest case encodes a single trait. By the word "phenotype", by the way, we should understand the totality of all the signs of the observed state (for example, eye color, shape of the lobe, tendency to diabetes, lactose digestion). And then the same question arises as in school years: what exactly needs to be read, is it possible to read not everything and how to deal with what has been read.
')
To make a mass medical service out of the technology of prognostic DNA tests, you need to pay attention to a few subtle points in data analysis. First, this is the problem of personalization: it is necessary to select markers carefully. Most studies aimed at establishing the genotype-phenotype association are conducted on large samples. Samples (analyzed groups of people), as a rule, are not very heterogeneous : they can be voluminous, but consist of representatives of one ethnic group. On the one hand, this simplifies the statistical analysis within the framework of the study, on the other hand, it raises the question of the relevance of the detected association in relation to other groups of people.
The second problem is the number of markers - the very case when you need to find a middle ground. And here we can not do without the eternal balancing between the two test parameters: sensitivity and specificity. An increase in the number of genetic markers undoubtedly increases the sensitivity of the assay to certain diseases. And at the same time, the specificity of the test may fall. Since we are talking about, for example, identifying a predisposition to serious diseases, a false positive error in identifying a terrible diagnosis will be more dangerous than specifying the likelihood of developing a disease. In addition, an increase in the number of markers leads to an increase in the cost of the test system, which also complicates access to the mass market.
There is no universal solution for these problems. At the initial stage of work, the researcher is faced with a situation where “there are a lot of articles, and they all need to be read.” For this reason, Genotek’s science department has proposed a selection algorithm that greatly simplifies the selection of polymorphisms for trait analysis. It is important that we are talking about polymorphisms, not about the genes entirely: two men have the same androgen receptor gene on each of the X chromosomes, but the first man has adenines (AA) in the rs6152 locus and the guanines in the second (GG). The likelihood that the last head of hair by 40 years will remain as lush is about 30% . In this case, to understand this, you do not need to read the entire gene sequence - just find and read only one point on the DNA, and then compare it with the one opposite.
What to look for
Geneticists call phenotypic peculiarities that we adopt from our ancestors. The fundamental problem of biology as a whole is the relationship between genotype and phenotype, as well as how one encodes the other. In our case, there are two views on the heritability of phenotypic features. On the one hand, the nature of inheritance may differ. Thus, a single trait may not be inherited at all, and its manifestation will in no way be associated with a genetic contribution. Such signs arising under the influence of lifestyle or external environmental conditions are of no interest to the developers of DNA tests.
On the other hand, if the trait is inherited, then it is important to understand what kind of genetic interaction underlies the observed state. In the simplest case, only one gene can influence the development of the disease - then we are talking about monogenic inheritance. For example, phenylketonuria is a metabolic disease that develops due to a “breakdown” in one gene. In this case, you can not talk about predisposition: if a person already has two copies of a broken gene, he will still develop the disease. However, in such a situation, genetic diagnosis can clarify the diagnosis for choosing a more adequate treatment.
It is more difficult when several genes contribute to the phenotypic feature. It is important to understand how the formation of the trait takes place, whether it is polygenic or multifactorial. Polygenic trait develops as a result of several polymorphisms - there is also no probabilistic nature of the development of the trait. With a certain combination of polymorphisms, you can reliably get a certain color of human eyes. Now the problem of predicting eye color (and other external features) is in forensic science. It is solved both with the help of mathematical models, and by training neural networks on large data sets. The multifactor phenotype, in addition to the genetic basis, develops under the influence of certain environmental conditions. These signs are of the greatest interest for medical prognostic analysis, since these are conditions that can be corrected with the help of a certain lifestyle.
The algorithm itself looks like this:
The first stage is a check for the presence of a genome wide association study (GWAS). This line of genetic research has been actively used with the development of microchipping technology. The purpose of the study is to find differences between the genomes of people with and without a specific trait. GWAS studies are characterized by large samples. For example, about 300,000 people took part in the GIANT (Genome Investigation of Antropometric Traits - a genomic study of anthropometric traits). The study confirmed the serious influence of the genetic factor on the development of obesity. The probability of observing the genotype-trait association in patients and healthy subjects, provided the association hypothesis is incorrect, is the p-value of the experiment. If this GWAS study value is less than 0.01 (adjusted for multiplicity), the polymorphism is placed on the short list of disease markers. If it is in the range of 0.01–0.05, compliance with one of the criteria of functional significance is required.
Criterion one: the direct or indirect mechanism of the influence of polymorphism on the trait is known. If the metabolic pathway of a substance is known (that is, the chain of transformations of substance A into substance X), then knowledge of which enzymes are involved in this chain may indicate the relevance of this polymorphism. For example, we know an enzyme that activates folic acid to catalyze the conversion of homocysteine to methionine. Methionine is an essential amino acid, and the accumulation of homocysteine can damage the endothelium of blood vessels. That is, we can talk about the replacement in the locus that encodes the active center of the enzyme, as the reason for the decline in homocysteine metabolism. Moreover, the presence of a single polymorphism of this kind does not guarantee the development of the disease.
If, according to the GWAS results, the value of the p-value of the association is greater than 0.05, the meta-analysis data are required. Meta-analysis is a study in which an association was identified by summarizing the results of a multitude of studies. If the meta-analysis shows the association “genotype-phenotype,” polymorphism is taken into work. If there is no data on meta-analysis, polymorphism is tested again by the criterion of functional significance.
Since the bulk of the work is the study of publications, there are strict criteria for the selection of scientific articles to find associations. For GWAS studies of these criteria 3:
- sample size - at least 750 patients in the primary study;
- P-value <0.01;
- associations must be confirmed in at least one study (not necessarily for rare diseases), and the impact factor of the publication must be at least 2.
For publications aimed at identifying compliance with the criterion of functional significance, the criteria are as follows:
- data should be obtained from tissue examinations (intravital biopsy, autopsy material, postoperative material) or biological fluids of participants;
- associations must be experimentally obtained in the scientific publication in question;
- P-value <0.05;
- the sample sizes of participants should be large enough to allow detection of the association of genetic markers with certain frequencies of occurrence;
- If there are several publications that have examined the association of this genetic marker with the risk of developing a disease, then choose for analysis: a) later (for example, from two articles 2009 and 2015 - an article for 2015); b) the publication in which the study was conducted on a larger number of samples.
The results of the meta-analysis, if any, are in priority. The requirements for meta-analysis are also very high: the publication itself must carefully select the sources, among which are both confirming and disproving the association of polymorphism with the attribute. Sources of information and keywords for which the search was conducted should be indicated, and justification of the criteria for inclusion and exclusion of publications (sample size, articles in English only, demographic characteristics of participants, etc.) is required. Special attention is paid to the so-called analysis of the bias of individual publications by the method of a funnel graph or sensitivity analysis. When the marker is included in the analysis, we focus on which populations took part in the study.
Despite the fact that the technology itself has long existed on the market, this is not some kind of “routine practice” according to which tons of manuals are written. The DNA test pipeline is working, and this is one of its sections. We will gradually introduce you to each of its fragments.
Source: https://habr.com/ru/post/401619/
All Articles