New approach to building the tree of life

When the British morphologist George Jackson Mivart [St. George Jackson Mivart] published in 1865 one of the first evolutionary trees, he lacked supporting material. He built a tree - a branching map of various species of primates - with the help of a detailed analysis of the spines of animals. The second tree, created on the basis of comparison of limbs of animals, showed other related connections between primates, highlighting the problem of evolutionary biology that still exists today.
Almost 150 years later, scientists acquired a mountain of data to build the so-called phylogenetic trees , a modern version of the structure created by Mivart. Advances in DNA decoding technology and bioinformatics make it possible to compare sequences of hundreds of genes, and sometimes whole genomes, in different species, and create the tree of life with more detail than has ever been possible.
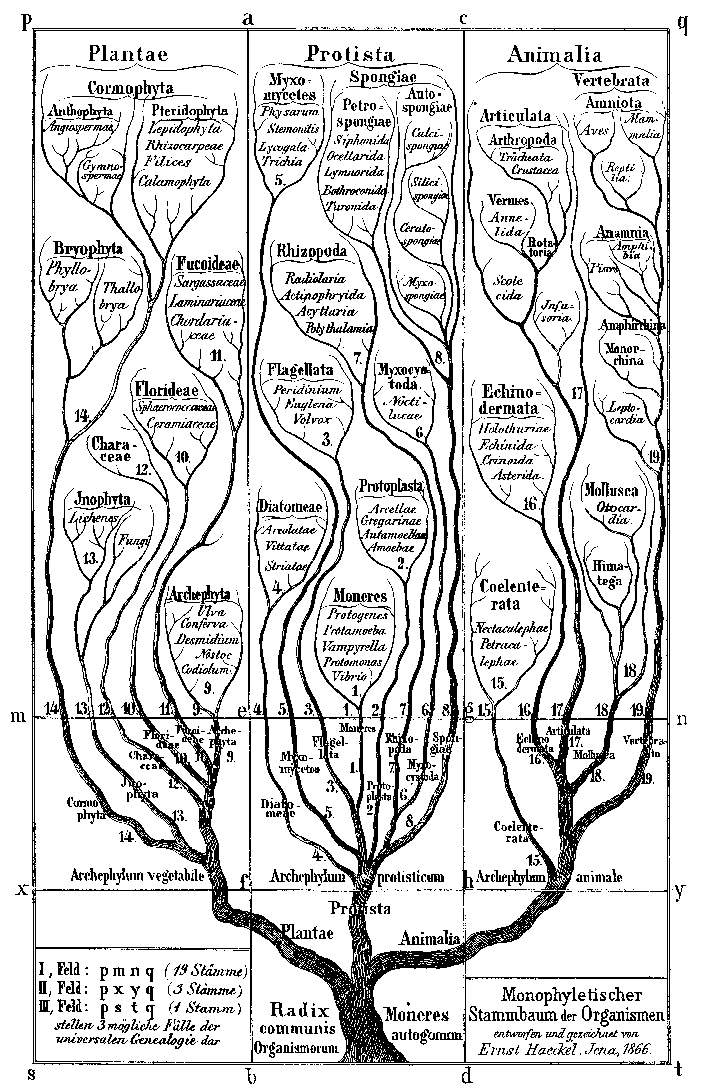
The historic tree of life from 1866 describes the kingdoms of plants, animals and single-celled
But although the abundance of data has helped to resolve some of the conflicts that have arisen over various sections of the evolutionary tree, it has brought new difficulties. The current version of the tree of life looks more like a controversial Wikipedia page than a published book — there are constant controversies about some of the branches. Just as the spine and limbs have led to the emergence of contradictory primate evolution maps, scientists now know that different genes in the same organism can tell different stories.
')
According to a new study, partly based on the study of yeast, the controversial picture drawn by individual genes is even more controversial than expected. "They claim that each of the 1070 genes is involved in a conflict," says Michael Donoghue , an evolutionary biologist at Yale, who is not associated with the study. “We are trying to understand the phylogenetic relationships of 1.8 million species, and we ourselves cannot sort out twenty types of yeast,” he says.
To resolve the paradox, researchers developed an algorithm based on information theory to measure the level of confidence in the correctness of individual parts of the tree. They hope that the new approach will help bring clarity to the periods of evolution, possessing both the most interesting and useful and the most conflicting data - for example, the Cambrian explosion - the rapid diversification of animal life that occurred 540 million years ago.
“Historically, the most interesting episodes are connected with the areas that attracted attention and caused controversy,” such as the origin of animals, vertebrates, and flowering plants, says Anthony Rokas , a biologist at Vanderbilt University who led the new study.
Based on the results of the new algorithm, scientists can choose only the most informative genes for the construction of phylogenetic trees. Such an approach can make the process both more accurate and efficient. “I think it will help speed up the reconstruction of the tree of life,” says Khidir Hilu, a biologist at the Virginia Institute of Technology.
Bricks of life
The basis of phylogenetic trees is created through the grouping of species according to their degree of kinship. If we compare the DNA of humans, chimpanzees and fish, it becomes clear that humans and chimpanzees are closer to each other than to fish.
Researchers once used one or more genes to compare organisms. But in the last decade there has been an explosion of phylogenetic data that very quickly filled the bases necessary for the creation of these trees. The analysis made it possible to fill in a few of the white spots scattered around the tree, but serious disagreements still exist.
For example, it is still unclear whether snails are closer in relation to them - bivalve mollusks or shovel-legged mollusks , Rokas says. It is not known how exactly one of the earliest branches of animals from a tree are connected, such as jellyfish and sponges. Scientists can demonstrate examples of conflicting trees that appear in the same scientific journals with a difference of weeks, or even in the same issue .
“Hence the question: why is it so difficult for us to come to an agreement?” Rokas says.

Rokas and his graduate student Leonidas Salichos studied this issue by evaluating the genes separately , using the most useful genes — carrying the most information related to evolutionary history — to build their version of the tree.
They started with 23 yeast species and selected 1070 genes. To begin with, they created a phylogenetic tree in a standard way, by concatenation. To do this, all the sequences from individual species come together in one megagene, and then the sequences of individual species are compared with this long sequence, on the basis of which a tree is created that best explains the differences.
The resulting tree is accurate in terms of standard statistical analysis. But since similar methods lead to the appearance of trees full of disagreements, Rokas and Salichos decided to delve into the subject. They built sets of phylogenetic trees for individual yeast genes, and applied an algorithm, developed using information theory, to search for the areas of greatest fit between different trees. The result, published in the journal Nature in May , was unexpected. It seems that each gene studied tells a slightly different evolutionary history.
“Virtually all trees built for individual genes were in conflict with a tree based on a concatenation of data,” says Hilu. “This is shocking.”
They concluded that if several genes support a particular architecture, then it must be exact. But if different sets of genes equally support two different architectures, then the probability of their exact correspondence to reality decreases. Rokas and Salichos used a method called statistical bootstrap to select the most informative genes.
In fact, “if you take only genes with active support, then you will get the right tree,” says Donogue.
The revised tree coincided with a tree built on an alternative source of evolutionary information — large-scale changes in DNA segments passed down from generation to generation — which justified their research.
Discoveries are not limited to yeast. By applying the same analysis to larger and more complex forms of life, including the genetic data of vertebrates and animals, they found serious conflicts between individual genes.
Some researchers need to get used to the idea of selectively excluding data from analysis. “For many years, the overriding problem of people trying to understand the relationships of organisms was the problem of collecting enough data,” said Jeffrey Townsend , an evolutionary biologist from Yale University who is not associated with research. “The community has always been told about the need for a data set, so it’s not surprising that they approached the task in this way.”
Although evolutionary biologists have struggled with these problems for years, the new study was the largest attempt to study the level of conflict of individual genes. “People will have two reactions: there are more conflicts than I thought, and we need to learn how to analyze them better,” says Donague, who wants to apply the new method in his work. However, he also points to the difficulties in confirming the accuracy of the new approach. Although the revised tree coincides with that built on alternative genetic information, the latter may reveal its own inconsistencies. “I'm not sure that we know what the relationship really is,” he says. “And if we are not sure about the true state of things, we don’t know if we got the right tree.”
Changing picture
Researchers need to apply the new technique more broadly to see how it can change the concept of evolution. However, Rokas and Salichos have already shown that it is most difficult to reconstruct short branches of the tree, or “bushy” parts of it, representing periods of rapid speciation — especially those closest to the base of the tree and deep in the evolutionary history.
“Theoretical studies predicted this behavior, but our study demonstrates confirmation for the first time with experimental data,” Rokas said.
Rokas argues that the new discoveries will change how researchers interpret fuzzy fragments of the tree. “Evolutionary biologists usually assume that if the tree doesn’t have the necessary details, then it’s wrong. And consequently, if we collect more data and compose better algorithms, then we will come to the correct tree, ”he says. But the presence of conflicting parts of the tree that persist, despite the flow of data and the use of a new type of analysis, may indicate the presence of bushy parts. "I think in some cases the algorithm will be able to resolve this conflict, and in others it will mark the areas of conflict that we can hardly ever resolve."
The study of these bushy parts of the tree can give a new look at particularly interesting stages of evolution, for example, the Cambrian explosion, when life has moved from the predominance of simple organisms to a diverse set of animal species.
Other scientists agree that discoveries may affect how experts cope with conflicting ideas about evolution. “I think this is a harbinger of a paradigm shift,” said Townsend. “If we use suitable methods, we have the opportunity to learn more about the issues that have tormented us for a long time.”
Townsend, who developed his own method of selecting the most informative genes based on the speed of their evolution , notes that not all members of the scientific community agree on the need for new approaches. “I hope this work will help bring this problem to the forefront,” he said.
Choosing the right number of genes to build prototypes of phylogenetic trees is not the only question that torments evolutionary biologists. They also need to agree on how many species to include in processing - the more species in the tree, the more difficult the analysis. Results may also differ due to differences in the quality of data collected for different species. “If we need to get a true evolutionary history of how everything is connected with each other, then what is better for this - collecting more genes or more species? - says Donogue. “I think both.”
New approaches that allow researchers to obtain accurate results using fewer genes may allow an evolutionary tree to expand. The ability to select only the most informative of genes can make the process more efficient, and allow scientists to create accurate trees using less data and resources. “If we could choose several genes and get the same good tree as with the whole genome,” says Hilu, “we could build a much more detailed tree of life — at the genus level, or even at the species level — instead of being content skeleton of the most important offshoots. "
Source: https://habr.com/ru/post/401343/
All Articles