Genome search with Wolfram Language (Mathematica) and HadoopLink
Translation of the post by Paul-Jean Letourneau " Searching Genomes with Mathematica and HadoopLink ".
The code given in the article can be downloaded here .
Note: This post is written as a continuation of the post Big Data in Mathematica with HadoopLink .
Translator’s note : the author of this article refers to the whole genome of a certain structural element of living matter as a gene. This is somewhat different from standard definitions that are close in meaning, meaning either the entire set of genes of a particular species (Ridley, M. (2006). Genome. New York, NY: Harper Perennial), or the full set of genetic instructions that can be found in cell ( http://www.genome.gov/Glossary/index.cfm?id=90 ). In this post we will use the presentation of the author.
In my previous post, I described how to write MapReduce algorithms ( wikis ) in Mathematica using the HadoopLink package. Now let's dig a little deeper and write a more serious MapReduce algorithm.
I already wrote earlier about some entertaining possibilities in the field of genomics in Wolfram | Alpha . If you're interested, you can even search the human genome for specific DNA sequences. Biologists often need to find the location of the DNA fragment that they found in the laboratory to determine which animal the fragment belongs to or from which chromosome. Let's use HadoopLink to create a genomic search engine!
As before, we load the HadoopLink package:
')
And set the link on the main Hadoop node:

To illustrate the idea, take the small-sized human mitochondrial genome from GenomeData :

First we divide the genome into separate bases (A, T, C, G):

They will be our key-value pairs (k1, v1) . Values - the initial position of each base of the genome:
{k1, v1} = {base, position}
That is, we have just created an index for the genome.
Export this index to the Hadoop Distributed File System (HDFS):
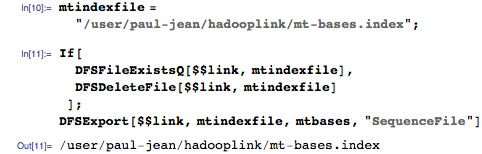
For queries, we will use a sequence of 11 bases about which we know that they are contained in the genome:

We use a sequence with a large number of repetitions in it in order to complicate the task of our algorithms.
Our genomic search engine for this query should return position 515:
Now we need a mapper and a reducer .
As it was said in the first part - mapper takes a key-value pair (step 1), and outputs another pair (step 2):

The mapper obtains a base from the genome index as an input value and returns key-value pairs for each location in the query, where base indices are found (you will soon find out why):
(1) Input: {k1, v1} = {base index, position in the genome}

The key at the exit is the position in the genome, the value at the exit is the position in the query:
(2) Conclusion: {k2, v2} = {position in the genome, position in the query}
What is the difference between the position in the genome and the position in the query? The position in the request is the position of the base in the request, while the position in the genome is the position in the whole genome.
For example, let's say mapper gets a key-value pair with base A at position 517:
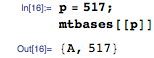
Positions in the request for base A in the requested GCACACACACA sequence are 3, 5, 7, 9, and 11:
Here is a sequence with highlighted positions:

Mapper has only one key-value pair with one index base along with the requested sequence. He does not have the rest of the genome to compare, so he needs to find all possible ways to build a query with base A at position 517:

Here, the colors correspond to each A in the query (horizontally) with their resulting positions in the genome (vertically). Take, for example, A in the third base in the query (shown in green). If it is inserted from A to position 517, then the requested sequence will begin in the genome from position 515 (517 - 3 + 1 = 515) (also shown in green).
In the same way, the highlighted red base (fifth position in the query) will force the requested sequence to line up, starting at position 513 in the genome (also shown in red). And the same for the seventh position in the query with 511 position in the genome, (purple) the ninth position in the query with 509 position in the genome (orange), the eleventh position in the query with 507 position in the genome (brown).
Only one of these arrangements is correct. In this case, for the third position in the request (green color), which allows the requested sequence to integrate into the genome. But the mapper does not know this, and therefore considers all possible matches.
So, the reducer collects the keys, it will collect all the bases that correspond to the same positions in the genome:
Input : {k2, {v2 ...}} = {position in the genome, {position in the query ...}}
Now for a given position in the genome, the reducer will look for those cases where the values form a complete sequence of query positions:

If the reducer finds full compliance, it gives the position of the genome:
Conclusion : {k3, v3} = {requested sequence, position in the genome}
Let's now request from our genomic search engine the sequence GCACACACACA:
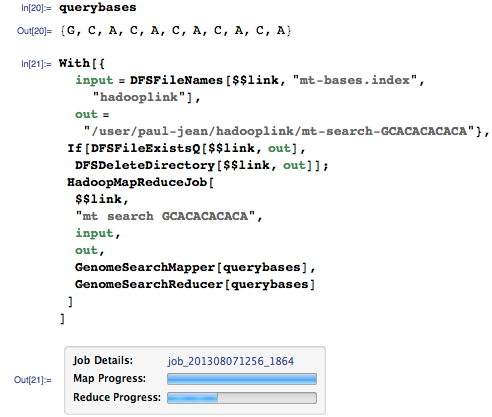
(See part 1 for a description of the HadoopMapReduceJob function).
And importing matches to the genome from HDFS:
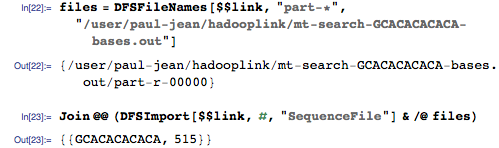
The corresponding position in the genome is 515, and this is the correct answer! Our genomic search engine works!
Let's now perform a search with a query to which two different positions in the genome must correspond:

This request must comply with position 10 and 2277:
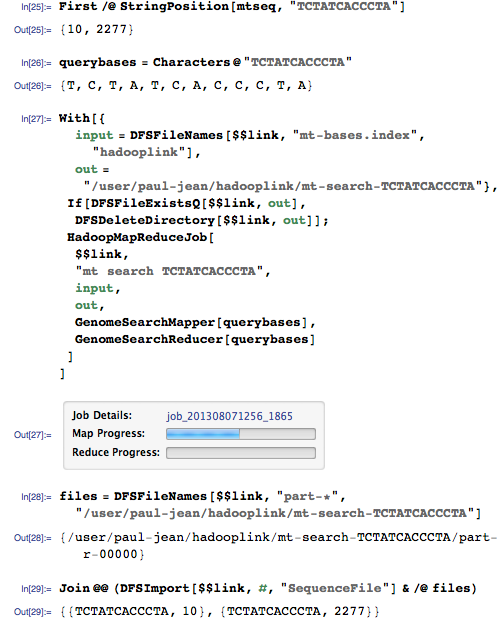
Yes, he found both coincidences!
Now let's scale it all for the entire human genome. The first step is to create an index, this time for the entire genome, not just the mitochondrial. To do this, I downloaded the entire human genome as text files from the state server and imported them into HDFS:
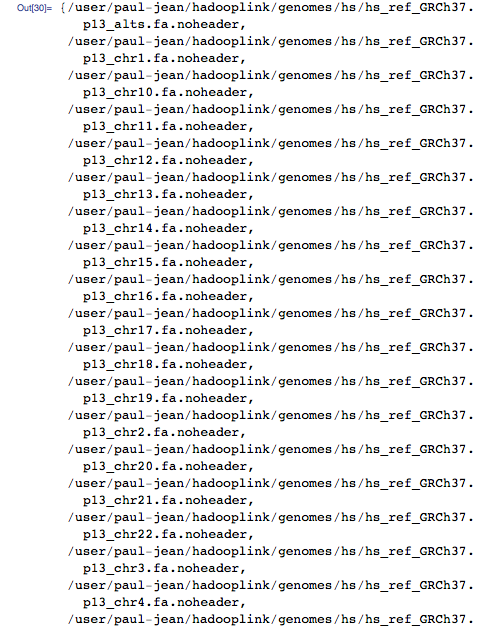

It turns out one text file per chromosome containing the raw chromosome sequences:
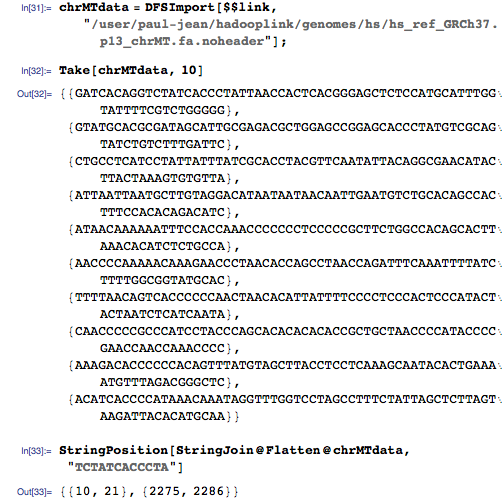
Then I applied MapReduce to create key-value pairs in the index on the HDFS, which looks like this:
[hs_ref_GRCh37.p13_alts.fa, 121] G
[hs_ref_GRCh37.p13_alts.fa, 122] A
[hs_ref_GRCh37.p13_alts.fa, 123] A
[hs_ref_GRCh37.p13_alts.fa, 124] T
[hs_ref_GRCh37.p13_alts.fa, 125] T
[hs_ref_GRCh37.p13_alts.fa, 126] C
[hs_ref_GRCh37.p13_alts.fa, 127] A
[hs_ref_GRCh37.p13_alts.fa, 128] G
[hs_ref_GRCh37.p13_alts.fa, 129] C
[hs_ref_GRCh37.p13_alts.fa, 130] T
One small difference from the previous example is that the key now is the {chromosome, position in the genome} , and the value is the basis on this position. So now the chromosome will be in the key. I changed the mapper slightly so that it could work with the new key view:

Reducer remained unchanged.
Let's run a search for the same sequence:
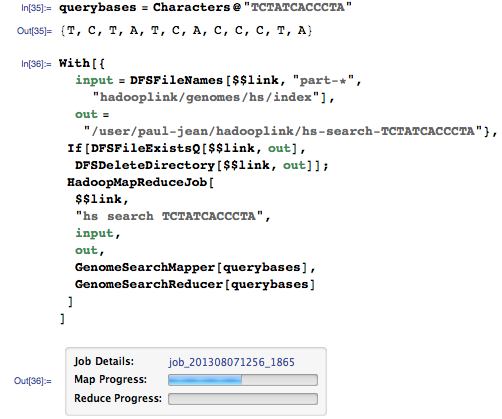
This time we get matches for the whole genome:
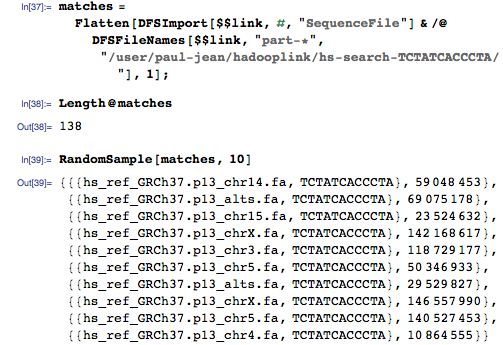
And we can still improve the algorithm. How about searching for inaccurate matches instead of exact ones? Slightly change the reducer , specifying how much of the request should match:

This is not the most efficient way to search the genome, but it does show how easy it is to write prototypes of MapReduce- based algorithms and run them in Mathematica. If you are interested in the details, I recommend to look at my recent posts . And you will be happy to answer your questions in HadoopLink GitHub Repo !
Source: https://habr.com/ru/post/263307/
All Articles