EPAM, get me a genome
If you compare a person with a computer, then his body is hardware, and what breathes life into it is software. And today we will talk about human software - its genome.

At present it is difficult to surprise someone with the terms "gene", "genome", "DNA", so tightly they entered into our daily lives. Everyone has heard that the human genome has been deciphered, but few of us clearly understand the significance of this scientific breakthrough for all of humanity. Correcting human “weaknesses”, increasing life expectancy and finding more and more effective ways to combat crime — all this becomes possible thanks to the study of the hereditary information contained in the human genome.
Since most of us have forgotten a little biology school course, we suggest refreshing the basic concepts:
')
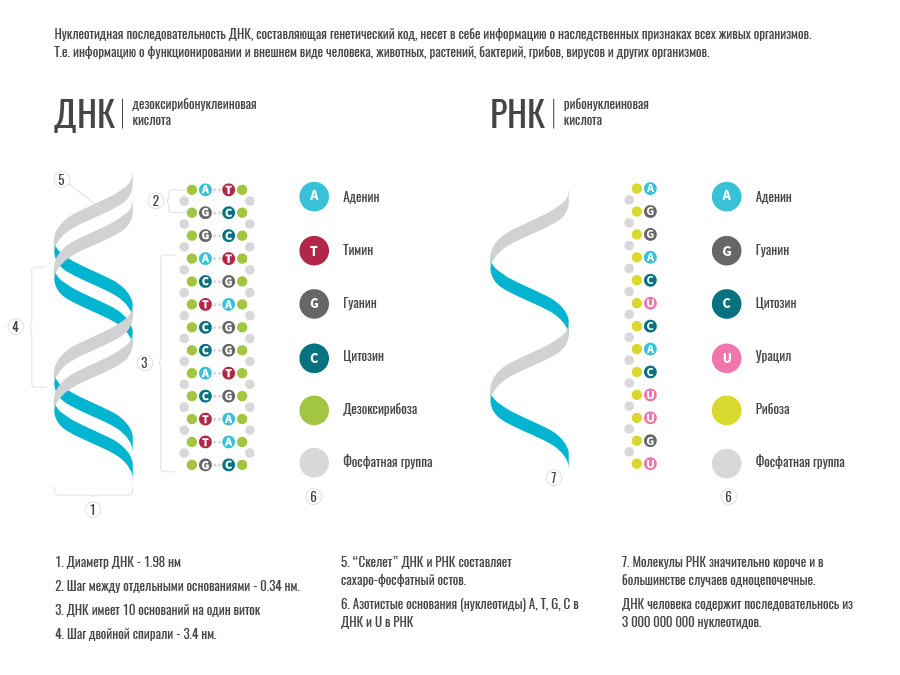
Deoxyribonucleic acid , better known as DNA , is a high-polymer natural compound contained in the nuclei of cells of all living organisms, the carrier of genetic information. DNA consists of 4 types of nucleotides. DNA nucleotides are based on deoxyribose sugar (the same for all nucleotides) and 4 types of nitrogenous bases - adenine (A), thymine (T), guanine (G) and cytosine. It is the nucleotide sequence that determines the information recorded in the DNA. Separate sections of DNA correspond to specific genes.
A gene , in turn, is a physical (specific DNA region) and functional (encodes a protein or ribonucleic acids) unit of heredity. The most important property of genes is the combination of their high resistance in a series of generations with the ability to inherit changes (mutations), which is the basis for the variability of organisms, which gives material for natural selection.
Ribonucleic acids (RNA) - a type of nucleic acids that consist of 4 nucleotides built on the basis of ribose sugar and nitrogenous bases - is adenine, guanine, cytosine, uracil (A, C, G, U). Uracil (U) is a complete analog of thymine (T) in DNA, so for our purposes we can consider them identical. In the cells of all living organisms, RNA is involved in the implementation of genetic information. An RNA copy is created from the genes located in the DNA, which either functions by itself or serves as a template for protein synthesis.
What is the genome? The genome is the DNA contained in the haploid set of chromosomes of a cell of a particular type of organism. In the expanded form, the genome is understood as the entire hereditary system of the cell.
About 23 pairs of chromosomes in the human genome, I think, everyone remembers. Initially, chromosomes were called structures of a very tightly folded filament, which forms in a cell at a certain point in the process of division, and is visible in an ordinary optical microscope. The chromosome is also called the DNA strand itself in the “expanded” state.
Reed is a separate reading of the DNA fragment. In turn, a locus is the location of a specific gene on a genetic or cytological map of a chromosome.
Everybody heard about the “decoding” of the human genome. Unfortunately, the term “decryption” was chosen poorly, which constantly raises questions. It is correct to say that the human genome was read (sequenced) , i.e., the complete nucleotide sequence of all DNA strands was obtained.
DNA reading is in itself a very difficult technical problem. But even after reading such a genome appears to us as a string consisting of four letters (A, C, G, T), and its length can reach several billion characters. At the same time, there are no absolutely clear “punctuation marks” in DNA that would indicate the beginning and end of sentence genes and other functionally significant elements. This huge amount of information is somewhat similar to a full computer memory dump. And to understand from this dump how our live computer works is a separate, much more difficult task.
Previously, only short pieces of the DNA sequence were available for analysis - individual genes and their fragments. Due to the complete sequencing of the genome, it has become much easier to analyze the “text” of DNA and look for functional areas in it. Over the past 10 years, scientists have made great strides in this direction. Were discovered new genes, mutant forms of which lead to cancer, atherosclerosis, Alzheimer's disease, cardiovascular diseases. A person's predisposition to alcoholism, drug addiction, gambling, mental illness and even suicide has a genetic basis. Moreover, the tendency to search for new impressions, maternal instinct, aggressive behavior, activity and irritability are also under strict genetic control! Of course, not every possibility inherent in DNA will be realized, but it is genes that give us a set of opportunities and risks, on the basis of which our life is built.
By the way, the traumatic experience of parents can be directly transmitted to descendants through the so-called epigenetic inheritance. The fears and stresses of the ancestors significantly influence the structure and function of the nervous system of subsequent generations. And if you are afraid of dogs for some reason, perhaps one of your ancestors was bitten. This area is still very little studied, but is also actively developing.
The DNA test allows you to detect a person's predisposition to certain diseases, including those caused by external causes (viruses, nutrition, etc.), to identify the person, establish paternity, determine the compatibility of the donor and recipient during transplantation, and much more.
Sometimes it happens. In 2002, American Lydia Fairchild, under divorce from her husband, underwent a DNA analysis procedure, which showed that she was not the mother of her two children. At that time, she was already pregnant with a third, whose blood samples after birth showed that she was not his mother. Further studies revealed that Lydia is a chimera, that is, an organism with two different genomes.
Complete sequencing of the individual’s genome allows you to simultaneously obtain the full text of DNA and analyze all its genes at once, instead of several thousand specialized tests for individual hereditary diseases. The project “Human Genome” demanded 5 years and several billion dollars. Modern machines make it possible to fully sequence the human genome in 15–20 days for about $ 1000 / genome. Then why is genome research and genomic diagnosis not happening as fast as we would like?
The fact is that no modern sequencer can read the entire DNA strand from beginning to end. For DNA sequencing, randomly cut into short fragments, which the sequencer then reads in parallel in large quantities. As a result, files of hundreds of gigabytes in size are obtained, in which there is an enormous number of short (150-300 nucleotides) readings.
Since there are many cells in one biological sample and DNA is fragmented randomly, the fragments read will overlap many times.
To obtain the desired genome, you need to assemble this puzzle in the full text. This is a huge computational task, and the processing of such files takes a long time. All data are processed by special genomic assemblers, which collect the longest sections of the genome.
The conventional wisdom that time is money is true for genomic research. Deciphering hereditary material opens up fantastic opportunities for medicine: from “repairing” defective genes to finding the secret of eternal youth. And that is why many scientific institutions and private companies are making incredible efforts to speed up the process of genome processing.
A large number of programs have been created for processing and analyzing the results of the sequence. However, sequencer performance and the amount of information received grow at such a rate that developers often do not have time to optimize their programs for processing an increasing amount of data. There is a need to adapt the developed programs for parallel work, the use of computing clusters, etc.
One of the clients turned to us for automating the process of controlling the quality of the assembly of the genome and reducing the computational time.
At the moment we are optimizing various processes to minimize time and, accordingly, costs. The work is carried out with small utilities that are engaged in the calculation of metrics — numerical characteristics by which the experimenter controls the quality of sample preparation, sequencing, and genome assembly. Metrics allow you to evaluate the file, which is issued sequencer: it is suitable for further analysis of the genome, or need to conduct another experiment.
Thanks to the work of our department of algorithms, the time to calculate one metric has been reduced from 11 hours to 105 minutes. Previously, four metrics serious car cheated for 25 hours. We, using multithreading and processing algorithms, reduced this time by four times. And this is not the limit.
The optimization of metrics consisted in finding points in the code for branching the computation process. All metrics are different in structure, but they have one thing in common: they all read a file in .bam format, which stores data on individual readings of DNA fragments (reads).
The optimization scheme is simple: data handlers use an iterator that reads one file from one file (SAMRecord) and analyzes the information with the accumulation of intermediate results (statistics) in some data structures.
One of the optimized metrics allows you to collect statistics from a .bam file, with which you can assess how well an experiment was conducted. To distinguish a sequencing error from a mutation, there should be a sufficiently large number of separate readings of DNA fragments (reads) in statistics. Very large coverage suggests that the experiment could be carried out cheaper without losing quality. As a result, the metric gives information about how many aligned grounds were filtered due to poor quality.
What is the optimization? Initially, the algorithm considered statistics for each location of a specific gene (locus), passing through all the reads covering this position. If the read consisted of 300 bases (and this is its standard length), then the algorithm read this DNA fragment 300 times to obtain information about the quality of its base. The algorithm, optimized by one of our specialists, reads the data on the quality of all the bases at the moment when the read is encountered for the first time, thus avoiding recourse to it for each locus. Information about the filtered bases accumulates in the array counter, which shifts as it passes through the genome. Thus, the statistics for each reading is collected only once, which allows you to significantly speed up the data processing.
Another metric collects statistics about duplicates (i.e., identical reads that match up to a given number of bases). Based on these statistics, the complexity of the read library is estimated. Too high number of duplicates shows that the experiment could be carried out cheaper. The low complexity of the library suggests that we are dealing with a small number of readings, which can distort the accuracy of the results with further work with the genome.
The original algorithm split reads into subsets, combining them by the first n-elements. Further, a comparison of readings of DNA fragments for each of them occurred. Obviously, such an algorithm works extremely slowly (for large subsets, time grows exponentially). The optimization in this case is the use of a modified LSH-algorithm ( locality-sensitive hashing algorithm ). Each reed is divided into shingles, that is, small pieces that include several bases and the position of the beginning of this piece in the read. Next, a table is built in which information is stored on which reads this shingle occurs in. Then, using random permutations, similar readings are determined, which are compared character by character. Using this optimization allows you to avoid a huge amount of unnecessary comparisons.
As mentioned above, our specialists were able to significantly reduce the time for calculating the metrics. This became possible not only due to the processing of algorithms, but also with the help of multithreading. What changes have been made?
A separate stream was created to read the records from the .bam file, which accumulates them in a separate circular buffer (bounded buffer). As soon as the buffer is filled, it is added up to a bounded queue. Both the buffer and the queue of such filled buffers are limited for reasons of the final amount of RAM allocated to the process.
Another stream (or several threads for some metrics) takes the next full buffer from this queue and uses it as a data source for the iterator. Then everything works the same as in the original scheme.
The accumulation of results from the processing of records from several streams requires the use of new data structures for statistics of metrics. This is done in order to avoid the race condition. To this end, we have created additional classes for the accumulation of statistics and apply atomic operations (atomics).
Atomic operations result in relatively high costs of consolidating intermediate statistical data from different streams for individual metrics. In order to speed up the processing, the records were made in the form of blocks of several hundreds in one stream.
Thanks to the optimization, the time for calculating the metrics has been reduced several times. However, this is not the limit. For sorted indexed .bam files, it is possible to switch to parallel reading and processing of data on individual chromosomes, which will reduce the processing time of genome data even more.
We can talk about genetics and the genome almost endlessly, since the topic is interesting and concerns us all. Perhaps due to the constant work on automating the processing of the genome, after just a couple of years, its decoding and assembly will take just a few hours, and the cost will decrease so much that absolutely everyone can afford to carry out a gene analysis. Fantasy? Hardly. Genomic technologies are now being developed almost according to Moore's law, the performance of sequencers doubles approximately every 2 years. Therefore, it is likely that over the next 10-15 years, genomic technologies will become as common and familiar as smartphones and laptops have become.
PS Due to the limited volume of the article does not pretend to strict scientific correctness in the description of biological terms and processes.
At present it is difficult to surprise someone with the terms "gene", "genome", "DNA", so tightly they entered into our daily lives. Everyone has heard that the human genome has been deciphered, but few of us clearly understand the significance of this scientific breakthrough for all of humanity. Correcting human “weaknesses”, increasing life expectancy and finding more and more effective ways to combat crime — all this becomes possible thanks to the study of the hereditary information contained in the human genome.
Some theory
Since most of us have forgotten a little biology school course, we suggest refreshing the basic concepts:
')
Deoxyribonucleic acid , better known as DNA , is a high-polymer natural compound contained in the nuclei of cells of all living organisms, the carrier of genetic information. DNA consists of 4 types of nucleotides. DNA nucleotides are based on deoxyribose sugar (the same for all nucleotides) and 4 types of nitrogenous bases - adenine (A), thymine (T), guanine (G) and cytosine. It is the nucleotide sequence that determines the information recorded in the DNA. Separate sections of DNA correspond to specific genes.
A gene , in turn, is a physical (specific DNA region) and functional (encodes a protein or ribonucleic acids) unit of heredity. The most important property of genes is the combination of their high resistance in a series of generations with the ability to inherit changes (mutations), which is the basis for the variability of organisms, which gives material for natural selection.
Ribonucleic acids (RNA) - a type of nucleic acids that consist of 4 nucleotides built on the basis of ribose sugar and nitrogenous bases - is adenine, guanine, cytosine, uracil (A, C, G, U). Uracil (U) is a complete analog of thymine (T) in DNA, so for our purposes we can consider them identical. In the cells of all living organisms, RNA is involved in the implementation of genetic information. An RNA copy is created from the genes located in the DNA, which either functions by itself or serves as a template for protein synthesis.
What is the genome? The genome is the DNA contained in the haploid set of chromosomes of a cell of a particular type of organism. In the expanded form, the genome is understood as the entire hereditary system of the cell.
About 23 pairs of chromosomes in the human genome, I think, everyone remembers. Initially, chromosomes were called structures of a very tightly folded filament, which forms in a cell at a certain point in the process of division, and is visible in an ordinary optical microscope. The chromosome is also called the DNA strand itself in the “expanded” state.
Reed is a separate reading of the DNA fragment. In turn, a locus is the location of a specific gene on a genetic or cytological map of a chromosome.
Genome and medicine
Everybody heard about the “decoding” of the human genome. Unfortunately, the term “decryption” was chosen poorly, which constantly raises questions. It is correct to say that the human genome was read (sequenced) , i.e., the complete nucleotide sequence of all DNA strands was obtained.
DNA reading is in itself a very difficult technical problem. But even after reading such a genome appears to us as a string consisting of four letters (A, C, G, T), and its length can reach several billion characters. At the same time, there are no absolutely clear “punctuation marks” in DNA that would indicate the beginning and end of sentence genes and other functionally significant elements. This huge amount of information is somewhat similar to a full computer memory dump. And to understand from this dump how our live computer works is a separate, much more difficult task.
Previously, only short pieces of the DNA sequence were available for analysis - individual genes and their fragments. Due to the complete sequencing of the genome, it has become much easier to analyze the “text” of DNA and look for functional areas in it. Over the past 10 years, scientists have made great strides in this direction. Were discovered new genes, mutant forms of which lead to cancer, atherosclerosis, Alzheimer's disease, cardiovascular diseases. A person's predisposition to alcoholism, drug addiction, gambling, mental illness and even suicide has a genetic basis. Moreover, the tendency to search for new impressions, maternal instinct, aggressive behavior, activity and irritability are also under strict genetic control! Of course, not every possibility inherent in DNA will be realized, but it is genes that give us a set of opportunities and risks, on the basis of which our life is built.
By the way, the traumatic experience of parents can be directly transmitted to descendants through the so-called epigenetic inheritance. The fears and stresses of the ancestors significantly influence the structure and function of the nervous system of subsequent generations. And if you are afraid of dogs for some reason, perhaps one of your ancestors was bitten. This area is still very little studied, but is also actively developing.
The DNA test allows you to detect a person's predisposition to certain diseases, including those caused by external causes (viruses, nutrition, etc.), to identify the person, establish paternity, determine the compatibility of the donor and recipient during transplantation, and much more.
Sometimes it happens. In 2002, American Lydia Fairchild, under divorce from her husband, underwent a DNA analysis procedure, which showed that she was not the mother of her two children. At that time, she was already pregnant with a third, whose blood samples after birth showed that she was not his mother. Further studies revealed that Lydia is a chimera, that is, an organism with two different genomes.
Genome and Bioinformatics
Complete sequencing of the individual’s genome allows you to simultaneously obtain the full text of DNA and analyze all its genes at once, instead of several thousand specialized tests for individual hereditary diseases. The project “Human Genome” demanded 5 years and several billion dollars. Modern machines make it possible to fully sequence the human genome in 15–20 days for about $ 1000 / genome. Then why is genome research and genomic diagnosis not happening as fast as we would like?
The fact is that no modern sequencer can read the entire DNA strand from beginning to end. For DNA sequencing, randomly cut into short fragments, which the sequencer then reads in parallel in large quantities. As a result, files of hundreds of gigabytes in size are obtained, in which there is an enormous number of short (150-300 nucleotides) readings.
Since there are many cells in one biological sample and DNA is fragmented randomly, the fragments read will overlap many times.
To obtain the desired genome, you need to assemble this puzzle in the full text. This is a huge computational task, and the processing of such files takes a long time. All data are processed by special genomic assemblers, which collect the longest sections of the genome.
EPAM, get me a genome
The conventional wisdom that time is money is true for genomic research. Deciphering hereditary material opens up fantastic opportunities for medicine: from “repairing” defective genes to finding the secret of eternal youth. And that is why many scientific institutions and private companies are making incredible efforts to speed up the process of genome processing.
A large number of programs have been created for processing and analyzing the results of the sequence. However, sequencer performance and the amount of information received grow at such a rate that developers often do not have time to optimize their programs for processing an increasing amount of data. There is a need to adapt the developed programs for parallel work, the use of computing clusters, etc.
One of the clients turned to us for automating the process of controlling the quality of the assembly of the genome and reducing the computational time.
At the moment we are optimizing various processes to minimize time and, accordingly, costs. The work is carried out with small utilities that are engaged in the calculation of metrics — numerical characteristics by which the experimenter controls the quality of sample preparation, sequencing, and genome assembly. Metrics allow you to evaluate the file, which is issued sequencer: it is suitable for further analysis of the genome, or need to conduct another experiment.
Metrics and algorithms - work optimization
Thanks to the work of our department of algorithms, the time to calculate one metric has been reduced from 11 hours to 105 minutes. Previously, four metrics serious car cheated for 25 hours. We, using multithreading and processing algorithms, reduced this time by four times. And this is not the limit.
The optimization of metrics consisted in finding points in the code for branching the computation process. All metrics are different in structure, but they have one thing in common: they all read a file in .bam format, which stores data on individual readings of DNA fragments (reads).
The optimization scheme is simple: data handlers use an iterator that reads one file from one file (SAMRecord) and analyzes the information with the accumulation of intermediate results (statistics) in some data structures.
One of the optimized metrics allows you to collect statistics from a .bam file, with which you can assess how well an experiment was conducted. To distinguish a sequencing error from a mutation, there should be a sufficiently large number of separate readings of DNA fragments (reads) in statistics. Very large coverage suggests that the experiment could be carried out cheaper without losing quality. As a result, the metric gives information about how many aligned grounds were filtered due to poor quality.
What is the optimization? Initially, the algorithm considered statistics for each location of a specific gene (locus), passing through all the reads covering this position. If the read consisted of 300 bases (and this is its standard length), then the algorithm read this DNA fragment 300 times to obtain information about the quality of its base. The algorithm, optimized by one of our specialists, reads the data on the quality of all the bases at the moment when the read is encountered for the first time, thus avoiding recourse to it for each locus. Information about the filtered bases accumulates in the array counter, which shifts as it passes through the genome. Thus, the statistics for each reading is collected only once, which allows you to significantly speed up the data processing.
Another metric collects statistics about duplicates (i.e., identical reads that match up to a given number of bases). Based on these statistics, the complexity of the read library is estimated. Too high number of duplicates shows that the experiment could be carried out cheaper. The low complexity of the library suggests that we are dealing with a small number of readings, which can distort the accuracy of the results with further work with the genome.
The original algorithm split reads into subsets, combining them by the first n-elements. Further, a comparison of readings of DNA fragments for each of them occurred. Obviously, such an algorithm works extremely slowly (for large subsets, time grows exponentially). The optimization in this case is the use of a modified LSH-algorithm ( locality-sensitive hashing algorithm ). Each reed is divided into shingles, that is, small pieces that include several bases and the position of the beginning of this piece in the read. Next, a table is built in which information is stored on which reads this shingle occurs in. Then, using random permutations, similar readings are determined, which are compared character by character. Using this optimization allows you to avoid a huge amount of unnecessary comparisons.
Multithreading
As mentioned above, our specialists were able to significantly reduce the time for calculating the metrics. This became possible not only due to the processing of algorithms, but also with the help of multithreading. What changes have been made?
A separate stream was created to read the records from the .bam file, which accumulates them in a separate circular buffer (bounded buffer). As soon as the buffer is filled, it is added up to a bounded queue. Both the buffer and the queue of such filled buffers are limited for reasons of the final amount of RAM allocated to the process.
Another stream (or several threads for some metrics) takes the next full buffer from this queue and uses it as a data source for the iterator. Then everything works the same as in the original scheme.
The accumulation of results from the processing of records from several streams requires the use of new data structures for statistics of metrics. This is done in order to avoid the race condition. To this end, we have created additional classes for the accumulation of statistics and apply atomic operations (atomics).
Atomic operations result in relatively high costs of consolidating intermediate statistical data from different streams for individual metrics. In order to speed up the processing, the records were made in the form of blocks of several hundreds in one stream.
Thanks to the optimization, the time for calculating the metrics has been reduced several times. However, this is not the limit. For sorted indexed .bam files, it is possible to switch to parallel reading and processing of data on individual chromosomes, which will reduce the processing time of genome data even more.
At last
We can talk about genetics and the genome almost endlessly, since the topic is interesting and concerns us all. Perhaps due to the constant work on automating the processing of the genome, after just a couple of years, its decoding and assembly will take just a few hours, and the cost will decrease so much that absolutely everyone can afford to carry out a gene analysis. Fantasy? Hardly. Genomic technologies are now being developed almost according to Moore's law, the performance of sequencers doubles approximately every 2 years. Therefore, it is likely that over the next 10-15 years, genomic technologies will become as common and familiar as smartphones and laptops have become.
PS Due to the limited volume of the article does not pretend to strict scientific correctness in the description of biological terms and processes.
Source: https://habr.com/ru/post/257215/
All Articles