Finishing the genome: fast, high quality, inexpensive
I think that many Habr readers have already heard about bioinformatics, perhaps even directly about the task of assembling the genome . Many people around the world are busy writing genomic assemblers - programs that interpret the raw data of machines for sequencing and produce the resulting DNA sequence of the studied organism. However, in most cases, the whole genome "out of the box" can not be obtained. In this article I will try to explain why the genome cannot be assembled with one click of the mouse and describe the process of its “finishing” - perhaps the most time-consuming step in the entire assembly, sometimes lasting several years.
Also, I will tell you how we can sometimes significantly facilitate this process, using the already collected genomes of closely related organisms. I was engaged in this task within the framework of writing my master's thesis at the St. Petersburg Academic University , and I studied in conjunction with the Institute of Bioinformatics . Since the resulting algorithm is quite specific, I will begin by describing the problem as a whole, I will give an overview of some of the "hardwired" methods for solving it, and then talk a little about what happened to me.
So we want to assemble the genome. On Habré there are already a couple of good publications on the topic of genomic assemblers: one , two .
If bioinformatics is something new for you, I highly recommend starting over with them, since this article implies knowledge of some basic concepts.
')
Just in case, let me remind you that a genomic assembler (collector) is a program that accepts short (several hundred nucleotides) overlapping pieces of the genome, called reads, and assembles them into a single sequence. More precisely, it tries to assemble - in most cases, even for relatively small bacterial genomes (several million nucleotides), the whole sequence does not work. This happens for a number of reasons, described in the articles above. I will briefly say that this happens because of the presence of repetitive pieces in the genome, areas that are physically difficult to read, as well as errors in the sequencing process.

As a result, instead of giving us the whole genome, we get its pieces, called contigs. They are already much longer (tens and hundreds of thousands of nucleotides), and in their totality they make up the original sequence. However, their correct order is unknown to us. What to do next?
To begin with, let us ask ourselves the question: why do we need a whole genome? After all, the length of contigs is enough, for example, to search for genes inside them. On the other hand, some genes will still be damaged due to fragmentation. But, what is more important, for some studies it is the structure of the genome that is necessary the order of the studied genes in it.
For example, this is important when studying evolution. As we well know, DNA mutates over time. Most often, small changes occur: one nucleotide is replaced with another, or several consecutive nucleotides are cut / inserted into another place. But there are much more rare, but serious changes. For example, a large piece of the genome can roll over or "move" to another position. It happens that whole chromosomes merge with each other, or vice versa, they break up into two parts. In their work, Hannehalli and Pevzner showed that the human and mouse genomes are separating a total of 131 such rearrangements. Agree, not so much.
Now that I am interested in you, let's get this genome together! We have a set of contigs - its pieces, the order of which is unknown to us. What will we do next? We can be helped by the technology of targeted sequencing, which allows one to read a specific and relatively short (up to several thousand nucleotides) region of the genome containing a specific pattern at the ends. For this, polymerase chain reaction ( PCR ) is used, and then the resulting product is read by a much more expensive, but reliable, sequencing method, the Sanger method . By running such a reaction and selecting the ends of two contigs of interest as a pattern, we can find out if they are adjacent in the genome, and also read the missing sequence between them. If our assumption is not true, and the contigs are too far apart, the reaction simply will not work.
But after all, we have hundreds (if we collect a bacterium) and thousands (mammals) of Contigs, respectively tens of thousands and millions of their possible "compounds." There is not enough time or money to check each of them. What do we do in this situation? Maybe we can somehow assess which compounds are most likely to exist, and which ones should not even be checked?
Of course we can! There are various methods (some of which I will now discuss) that allow you to combine contigs into the so-called scaffold, which is their ordered set. There are gaps of a certain length between contigs, symbolizing an unknown sequence:

It is clear that having such a “draft” of the genome, it will be much easier for us to fill in the gaps in it. Let's consider what are the ways of combining contigs into scaffolds (this process is called scaffolding).
What could be better than a short read? Yes, unless two are the same! Let's cut the DNA that we have to sequence into pieces of some known length, and then read each one from the beginning and end. Of course, most of the sequence between the ends will remain unread, but now we have a couple of readings, the distance between which we know.
Such pairs of reads give us new additional information. Let's "align" (align) them (that is, find an entry, possibly inaccurate) on our contigs. If at the same time two reads are aligned to different contigs, then the latter can be combined into a single scaffold:
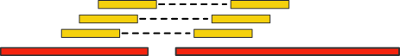
An important criterion of this technology is the distance between pairs of reads - the so-called insertion length. The larger it is, the longer we can pass over the longer genome intervals. However, with an increase in the length of the insert, the cost of the experiment increases significantly, as well as the number of errors.
Recently, technologies have begun to emerge that make it possible to produce much longer reads (up to several tens or even hundreds of thousands of base pairs) in relatively large quantities. For example, this opportunity provides technology PacBio . However, in all of them so far there are two significant drawbacks: firstly, the high cost of the process, and secondly, the presence of a large number of errors.
Collecting a genome with so many inaccuracies is another task. Some went the other way - the creation of hybrid assemblies. They combine the usual short reads, as well as long, but less accurate. The idea is intuitively clear: at first, as usual, we perform the assembly using only short reads, and then align the resulting contigs with long reads. This, as in the case of pair reads, gives us information about the location of contigs relative to each other. Also, the part of the genome between them becomes available:
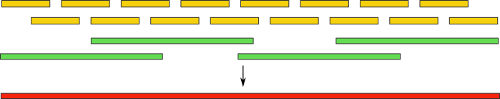
Using such a technique, not so long ago a group of scientists was able to fully automatically assemble the bacterial genome . Also, there is a noticeable success in the assembly using long reads alone. However, the whole technology, unfortunately, remains the lot of the elect because of its high cost and low prevalence.
Hi-C is a very fresh and promising technology that allows you to measure the interaction of pieces of the genome in space. As you probably know, DNA is not just a long linear molecule, it is also packed in a complex way into a spatial structure, which is called a chromosome. Without going into details, I will say that Hi-C allows you to get pairs of reads for which the corresponding pieces of the genome are close in space, but they are not necessarily close if we stretch the entire molecule in one line. The picture below shows the intensity of the interaction of chromosome segments with each other (along both axes - linear coordinates on the chromosome):

So we can get quite interesting information reflecting the form of DNA packaging in the cell. How can this be used for scaffolding? It's very simple: pieces of DNA that are linearly close to each other will, on average, more often interact with each other (which we see in the picture above). Again, I will not indulge in long explanations and refer the reader to the original article on this topic.
Finally, we very smoothly approached the topic of my work. Since the advent of the first sequencing technologies, mankind has managed to accumulate quite a lot of data. There are already a large number of fully (or almost completely) assembled genomes of various organisms. Why not take advantage of this information, instead of carrying out expensive experiments?
And you can use! There are many purely computational methods under the common name “reference-assisted assembly”, which involve the use of sequences of already assembled related organisms to improve the quality of the assembly of the new sample. The basic idea here is similar to the one used for assembly with long reads: we find the alignment of the contigs now to the reference genome, and combine them in the appropriate order:
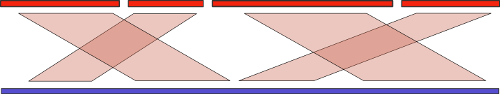
But, as we know, evolution does not stand still, and the differences in the collected and reference genome can be significant. First, it can lead to difficulties when searching for alignment. But it's not so bad. At the very beginning of the article, I mentioned large-scale genomic rearrangements. It is clear that if such rearrangements between the collected and reference genome are present, then we risk getting a completely wrong result. So, we need an algorithm that analyzes such restructuring and tries to "put everything in its place."
I was writing this algorithm as my master's thesis. Its main difference from existing solutions is the use of several reference genomes simultaneously. This gives us additional information about how the genomes have changed in the course of evolution, and therefore allows us to more reliably assess the order of the Contigs in our genome.
Without going into details, I will try to explain why several references are better than one. Let's start with the fact that between all the genomes that are given to us at the entrance, there is some kind of evolutionary relationship. Simply put, we can build a phylogenetic tree , in the leaves of which our genomes will be located, both reference - R and target - T. The tree shows their directed evolution from some common ancestor, and the length of the branches corresponds to the duration of the processes:

Now, knowing the structure of the reference genomes, we need to restore the target. To do this, it is necessary to understand how the evolution of the whole family proceeded. It is important for us to predict in which places of the phylogenetic tree genomic rearrangements took place, the consequences of which we observe in the leaves. Knowing this, we will be able to determine which reorganizations occurred before, and which after the separation of the genome we are collecting into a separate branch. So, we can and say which of them is most likely present in our assembly.
According to the results of the article was written, which became my master's thesis. The program itself can be found on the githaba - fenderglass.imtqy.com/Ragout . A detailed description of the algorithm, unfortunately, goes beyond the publication of the Habr format, and it would not be too clear without an additional background. But if this article arouses the interest of the community, then in the future I will continue to write about the bioinformatic algorithms associated with this topic, and I will gradually delve into the field.
PS I also wanted to write in Bioinformatics, but I lacked karma. - already wrote
Also, I will tell you how we can sometimes significantly facilitate this process, using the already collected genomes of closely related organisms. I was engaged in this task within the framework of writing my master's thesis at the St. Petersburg Academic University , and I studied in conjunction with the Institute of Bioinformatics . Since the resulting algorithm is quite specific, I will begin by describing the problem as a whole, I will give an overview of some of the "hardwired" methods for solving it, and then talk a little about what happened to me.
Introduction
So we want to assemble the genome. On Habré there are already a couple of good publications on the topic of genomic assemblers: one , two .
If bioinformatics is something new for you, I highly recommend starting over with them, since this article implies knowledge of some basic concepts.
')
Just in case, let me remind you that a genomic assembler (collector) is a program that accepts short (several hundred nucleotides) overlapping pieces of the genome, called reads, and assembles them into a single sequence. More precisely, it tries to assemble - in most cases, even for relatively small bacterial genomes (several million nucleotides), the whole sequence does not work. This happens for a number of reasons, described in the articles above. I will briefly say that this happens because of the presence of repetitive pieces in the genome, areas that are physically difficult to read, as well as errors in the sequencing process.
As a result, instead of giving us the whole genome, we get its pieces, called contigs. They are already much longer (tens and hundreds of thousands of nucleotides), and in their totality they make up the original sequence. However, their correct order is unknown to us. What to do next?
To begin with, let us ask ourselves the question: why do we need a whole genome? After all, the length of contigs is enough, for example, to search for genes inside them. On the other hand, some genes will still be damaged due to fragmentation. But, what is more important, for some studies it is the structure of the genome that is necessary the order of the studied genes in it.
For example, this is important when studying evolution. As we well know, DNA mutates over time. Most often, small changes occur: one nucleotide is replaced with another, or several consecutive nucleotides are cut / inserted into another place. But there are much more rare, but serious changes. For example, a large piece of the genome can roll over or "move" to another position. It happens that whole chromosomes merge with each other, or vice versa, they break up into two parts. In their work, Hannehalli and Pevzner showed that the human and mouse genomes are separating a total of 131 such rearrangements. Agree, not so much.
Genome draft
Now that I am interested in you, let's get this genome together! We have a set of contigs - its pieces, the order of which is unknown to us. What will we do next? We can be helped by the technology of targeted sequencing, which allows one to read a specific and relatively short (up to several thousand nucleotides) region of the genome containing a specific pattern at the ends. For this, polymerase chain reaction ( PCR ) is used, and then the resulting product is read by a much more expensive, but reliable, sequencing method, the Sanger method . By running such a reaction and selecting the ends of two contigs of interest as a pattern, we can find out if they are adjacent in the genome, and also read the missing sequence between them. If our assumption is not true, and the contigs are too far apart, the reaction simply will not work.
But after all, we have hundreds (if we collect a bacterium) and thousands (mammals) of Contigs, respectively tens of thousands and millions of their possible "compounds." There is not enough time or money to check each of them. What do we do in this situation? Maybe we can somehow assess which compounds are most likely to exist, and which ones should not even be checked?
Of course we can! There are various methods (some of which I will now discuss) that allow you to combine contigs into the so-called scaffold, which is their ordered set. There are gaps of a certain length between contigs, symbolizing an unknown sequence:
It is clear that having such a “draft” of the genome, it will be much easier for us to fill in the gaps in it. Let's consider what are the ways of combining contigs into scaffolds (this process is called scaffolding).
Scaffolding technology
Paired reading libraries
What could be better than a short read? Yes, unless two are the same! Let's cut the DNA that we have to sequence into pieces of some known length, and then read each one from the beginning and end. Of course, most of the sequence between the ends will remain unread, but now we have a couple of readings, the distance between which we know.
Such pairs of reads give us new additional information. Let's "align" (align) them (that is, find an entry, possibly inaccurate) on our contigs. If at the same time two reads are aligned to different contigs, then the latter can be combined into a single scaffold:
An important criterion of this technology is the distance between pairs of reads - the so-called insertion length. The larger it is, the longer we can pass over the longer genome intervals. However, with an increase in the length of the insert, the cost of the experiment increases significantly, as well as the number of errors.
Long reeds
Recently, technologies have begun to emerge that make it possible to produce much longer reads (up to several tens or even hundreds of thousands of base pairs) in relatively large quantities. For example, this opportunity provides technology PacBio . However, in all of them so far there are two significant drawbacks: firstly, the high cost of the process, and secondly, the presence of a large number of errors.
Collecting a genome with so many inaccuracies is another task. Some went the other way - the creation of hybrid assemblies. They combine the usual short reads, as well as long, but less accurate. The idea is intuitively clear: at first, as usual, we perform the assembly using only short reads, and then align the resulting contigs with long reads. This, as in the case of pair reads, gives us information about the location of contigs relative to each other. Also, the part of the genome between them becomes available:
Using such a technique, not so long ago a group of scientists was able to fully automatically assemble the bacterial genome . Also, there is a noticeable success in the assembly using long reads alone. However, the whole technology, unfortunately, remains the lot of the elect because of its high cost and low prevalence.
Hi-c
Hi-C is a very fresh and promising technology that allows you to measure the interaction of pieces of the genome in space. As you probably know, DNA is not just a long linear molecule, it is also packed in a complex way into a spatial structure, which is called a chromosome. Without going into details, I will say that Hi-C allows you to get pairs of reads for which the corresponding pieces of the genome are close in space, but they are not necessarily close if we stretch the entire molecule in one line. The picture below shows the intensity of the interaction of chromosome segments with each other (along both axes - linear coordinates on the chromosome):
So we can get quite interesting information reflecting the form of DNA packaging in the cell. How can this be used for scaffolding? It's very simple: pieces of DNA that are linearly close to each other will, on average, more often interact with each other (which we see in the picture above). Again, I will not indulge in long explanations and refer the reader to the original article on this topic.
Reference Assembly
Finally, we very smoothly approached the topic of my work. Since the advent of the first sequencing technologies, mankind has managed to accumulate quite a lot of data. There are already a large number of fully (or almost completely) assembled genomes of various organisms. Why not take advantage of this information, instead of carrying out expensive experiments?
And you can use! There are many purely computational methods under the common name “reference-assisted assembly”, which involve the use of sequences of already assembled related organisms to improve the quality of the assembly of the new sample. The basic idea here is similar to the one used for assembly with long reads: we find the alignment of the contigs now to the reference genome, and combine them in the appropriate order:
But, as we know, evolution does not stand still, and the differences in the collected and reference genome can be significant. First, it can lead to difficulties when searching for alignment. But it's not so bad. At the very beginning of the article, I mentioned large-scale genomic rearrangements. It is clear that if such rearrangements between the collected and reference genome are present, then we risk getting a completely wrong result. So, we need an algorithm that analyzes such restructuring and tries to "put everything in its place."
I was writing this algorithm as my master's thesis. Its main difference from existing solutions is the use of several reference genomes simultaneously. This gives us additional information about how the genomes have changed in the course of evolution, and therefore allows us to more reliably assess the order of the Contigs in our genome.
Without going into details, I will try to explain why several references are better than one. Let's start with the fact that between all the genomes that are given to us at the entrance, there is some kind of evolutionary relationship. Simply put, we can build a phylogenetic tree , in the leaves of which our genomes will be located, both reference - R and target - T. The tree shows their directed evolution from some common ancestor, and the length of the branches corresponds to the duration of the processes:
Now, knowing the structure of the reference genomes, we need to restore the target. To do this, it is necessary to understand how the evolution of the whole family proceeded. It is important for us to predict in which places of the phylogenetic tree genomic rearrangements took place, the consequences of which we observe in the leaves. Knowing this, we will be able to determine which reorganizations occurred before, and which after the separation of the genome we are collecting into a separate branch. So, we can and say which of them is most likely present in our assembly.
According to the results of the article was written, which became my master's thesis. The program itself can be found on the githaba - fenderglass.imtqy.com/Ragout . A detailed description of the algorithm, unfortunately, goes beyond the publication of the Habr format, and it would not be too clear without an additional background. But if this article arouses the interest of the community, then in the future I will continue to write about the bioinformatic algorithms associated with this topic, and I will gradually delve into the field.
Source: https://habr.com/ru/post/238759/
All Articles