MiTCR is a platform for diagnosing a new type. Yandex Workshop
The story of Dmitry Bolotin is dedicated to the MiTCR software, developed for the analysis of immunological receptor repertoires. In his report, he reviewed the main features of the analysis of raw sequencing data, in particular, sequence alignment algorithms and error correction in the source data, and also briefly described the architecture, performance, and the nearest development plan for the program. MiTCR source code is open. In the future, this software can lead to a common platform for bioinformatists, where they can process their data and share it with other researchers. The result of this collaboration should be a new type of diagnosis: with the help of a blood test, it will be possible to answer not only the question of whether a person has a particular disease, but immediately determine exactly what he is sick.

Video of the report
')
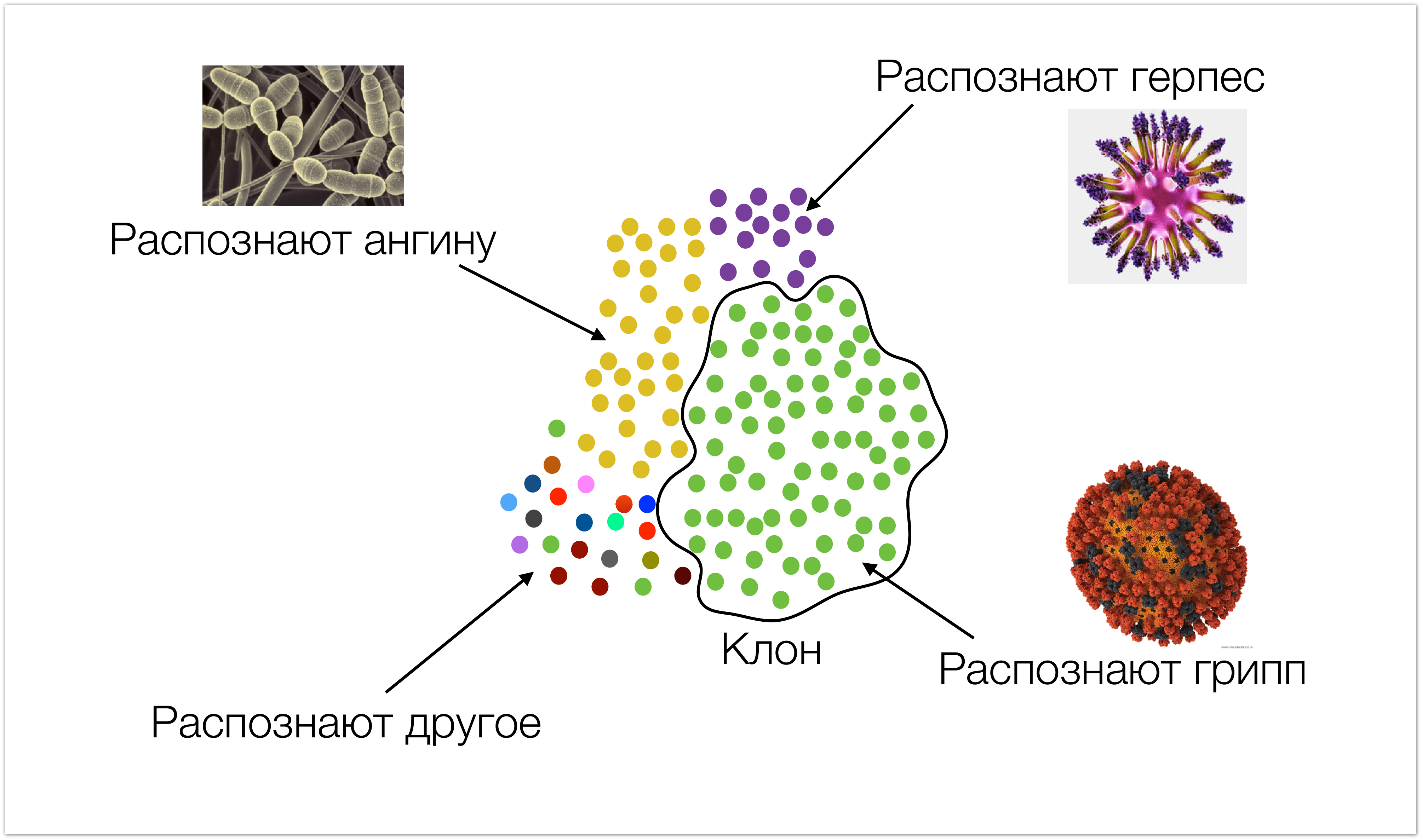
We will start from afar so that it is clear what data we are working with and where they come from. The picture under the cut is very schematically depicted immunity. Cells that have the same specificity are colored with the same color (i.e., they recognize the same types of infections). We call these cells clones. During an infection attack, the number of cells that recognize it increases.
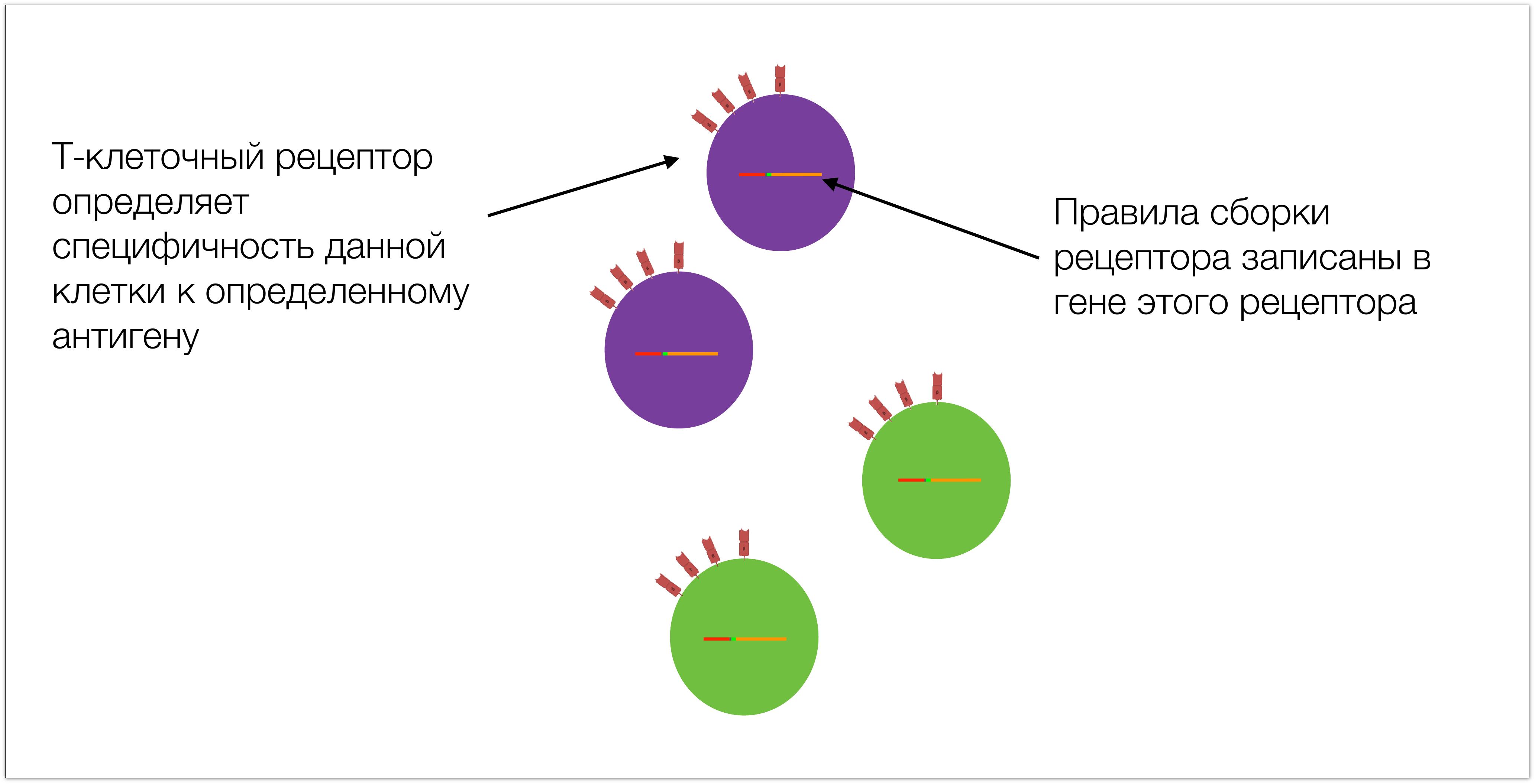
The specificity of these cells is due to the fact that they have a T-cell receptor on the surface, the assembly rules of which are recorded in the corresponding gene. For the subsequent narration, it is important to understand its structure.
A gene is a piece of DNA, you can think of it simply as a sequence consisting of a four-letter alphabet. The uniqueness of the T-cell receptor gene is that it is different in different cells. All other genes that are in our cells are the same, only the T cell receptor and antibody genes differ in T and B lymphocytes, respectively.
This gene consists of 4 main pieces. One piece in T-cell receptors is always the same. The other two sites (red and green in the picture) are selected from a small set. During the assembly of these two sections, random letters (about ten) are added between them, which leads to the formation of a giant variety of such receptors. The site that then recognizes an antigen in a protein is called CDR3.
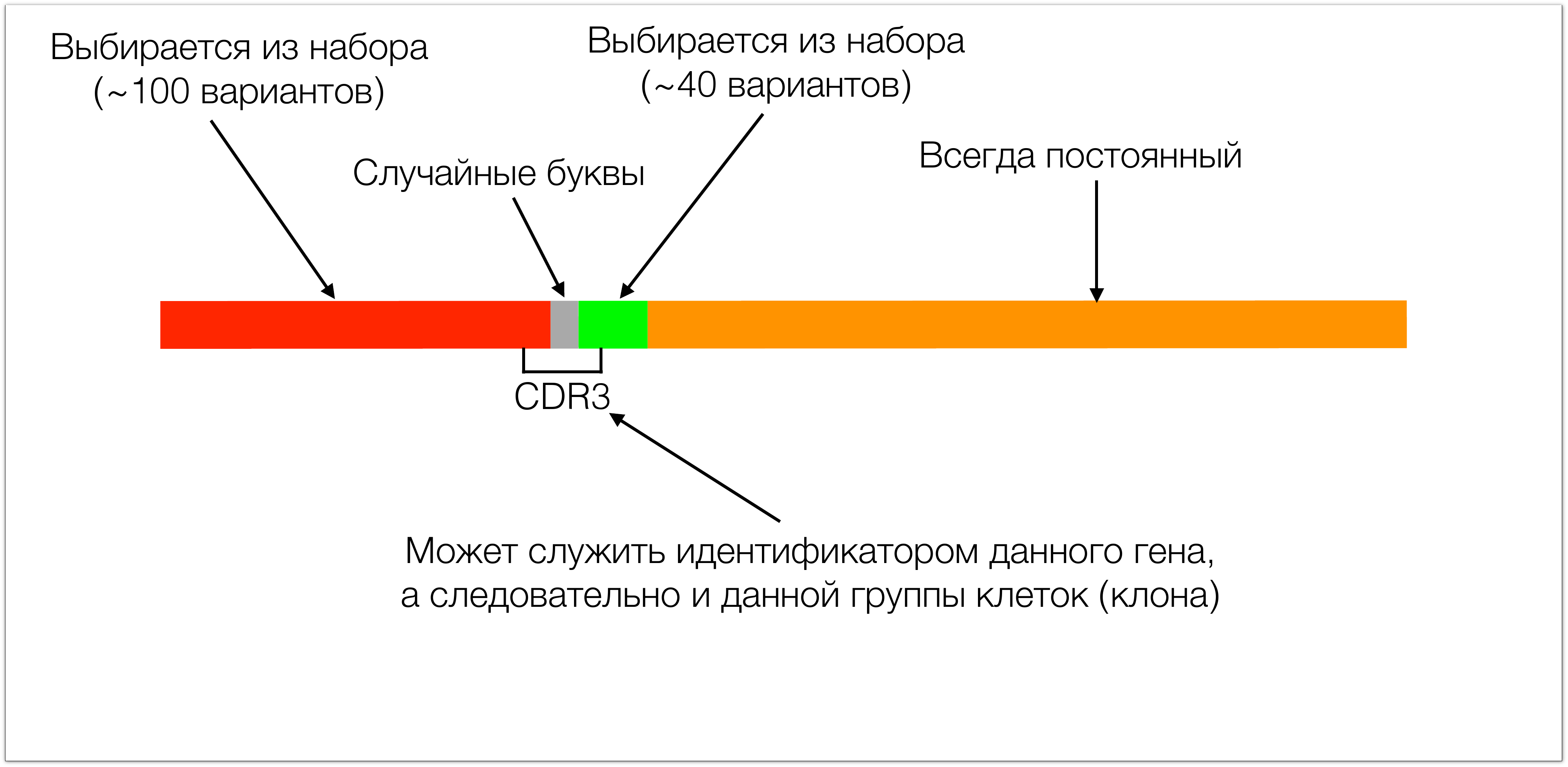
It is interesting from two sides: on the one hand, it determines the specificity, because simply by biology it just encodes the receptor site responsible for the recognition of antigen, and on the other hand it is convenient as an identifier, because all diversity is concentrated in it, it includes a piece of red plot, a piece of green. From these pieces we can fully determine which of the set of these segments was chosen, and all these random letters are fully concentrated in it. Therefore, knowing CDR3 we can reconstruct the entire protein. We can also use it as an identifier, because if CDR3 is the same, then the whole gene is also the same.
So, we have a blood sample, we carry out some sequence of complex molecular biological reactions in order to isolate from it DNA sequences containing CDR3. Next, we load them onto the sequencer, and at the output we get something like that:
These are real data that come to us from the sequencing center. If you discard too much, then in fact it is just DNA sequences from 100 to 250 nucleotides in length. And there are a lot of such sequences.
Our task is to reconstruct from this data what was at the very beginning find out how many and which clones were in the primary sample.
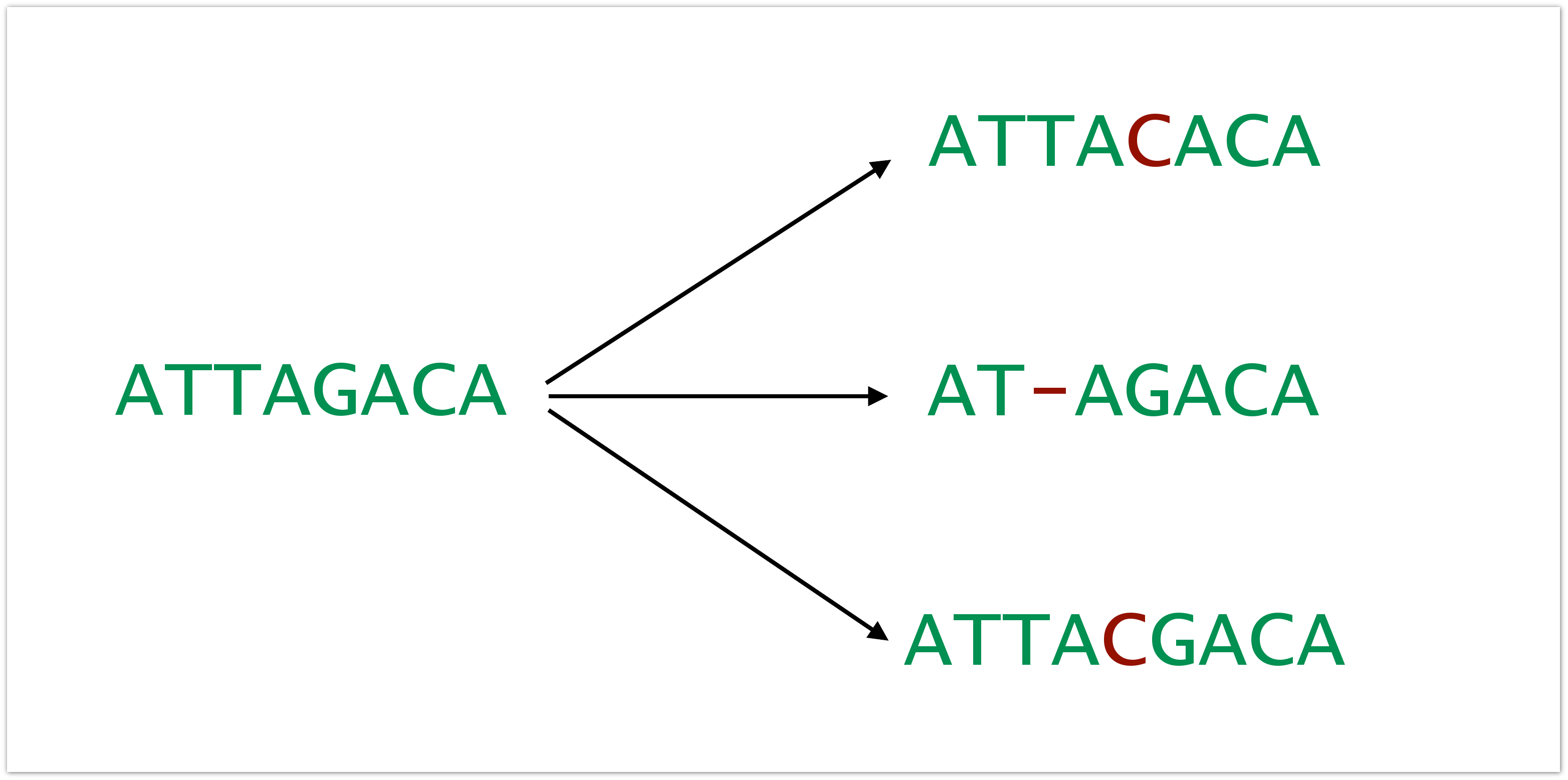
Everything is complicated by the fact that in the data there are a variety of errors. These errors are introduced at all stages: while sample preparation takes place, while it is sequenced, etc. The rate of error can vary, depending on the architecture of the experiment from the sequencer that was used, in general, from everything. These can be inserts, deletions and replacements.
And this is bad, first of all, from the point of view that the data on the sample are distorted and artificial diversity is introduced. Suppose we had a sample consisting of only one clone, but as a result of the preparation, mistakes were made to it. As a result, we see that we have more clones than there were, and we have the wrong picture of the patient's T-cell repertoire.
Now briefly about what data we work with. At the entrance, we get about a hundred million sequences, or even a billion, 100-250 nucleotides in length. All this in total weighs up to 100 GB. And the concentration of CDR3 it can be from 1/5 (in very rare cases it is more) and up to the read sequences are millions.
The scheme of MiCTR software developed by us looks approximately as follows:

MiCTR solves the problem I described earlier: it makes a list of clones from the data and corrects errors. The first block extracts from the CDR3 data, the next blocks work only with CDR3 already and combine them into clones and correct the errors.
On the first block I will not concentrate much. He is engaged in leveling, and this is a huge part of bioinformatics, which would require a separate lecture. In short, this block lays out the input sequences, determining where the red is in them and where the green areas are. The set from which they are selected is known, it is in the databases and absolutely the same for all people. We determine which of these red areas is found and where. Do the same with green areas. Having built these alignments, we already know where we have CDR3, and we can select them from the sequences and work with them.
The most trivial thing to do with this data is to combine them by the same: the same CDR3 must be collected and counted. But since there are errors in the data that need to be corrected, and to correct them, we will subsequently perform a fuzzy search on multiple CDR3s (for example, search for CDR3 that differ by one letter), then for storage we use a prefix tree (trie), which is very convenient for such operations . In addition, when growing, this tree is not rebuilt, so it is very convenient to implement all competitive algorithms on it. No need to worry much about any tricky syncs. This tree is very easy to use, but it has its drawbacks. It is heavy and weighs quite a lot in RAM. However, in the real world, it turns out that this does not pose a particular problem (it can be kept within 1-2), so this is an ideal container for such data.
What do we do in relation to errors? In fact, we collect all these clones from only a part of the sequences. The fact is that the sequencer, in addition to giving the letters that it has read, puts in each position a value of quality that determines the probability of error. Those. The sequencer read the letter, but he can be sure of it or not. And for each position is set the probability of error. And here we are doing a fairly simple trick: we just carry out a certain error probability threshold, say, one-hundredth. All the letters that have a high probability of error, we call bad. Accordingly, all CDR3 containing such a letter are also considered bad. All this is stored in a container that not only collects data by number, but also collects other information: it searches for equivalent CDR3, or CDR3 that differ only in positions with poor quality. Thus it is possible to correct a large proportion of errors. Unfortunately, it is impossible to get rid of all errors, some errors are made before the sequencer, so there is no additional information about them.
And here we are doing a fairly simple thing, we find CDR3 pairs that differ by one letter and are very different in concentration. Most often, this is a clear marker for errors. And we simply remove them, choosing a threshold of the order of 1 to 10 by concentration: one error per sequence.
In sum, all this allows us to correct 95% of the errors and not to remove the natural diversity. It is clear that with such algorithms you can make a mistake and take the natural clone for an error, but in fact they are so diverse that this does not happen. Such performance is a good number for real work.
It is worth a little to describe the software implementation of all this. The system is multi-threaded, the sequence markup is launched in parallel. In uniprocessor systems, the best result is achieved if the number of threads matches the number of cores. The diagram above shows work in two streams: one is engaged in the formation of clones from good CDR3, the other from bad ones. All this scales well: for example, on a six-core processor, the execution of the code is accelerated approximately 3.5-4 times. As for performance, but on a completely ordinary hardware (AMD Phenom II X4 @ 3.2 GHz) 50,000 sequences per second are processed. The storage of one clone type takes about 5 KB of RAM. Thus, the most complex (diverse) array from our experience was able to process in 20 minutes on a machine with the above processor and 16 GB of RAM.
Everything is written in Java, the source is open , there is a well-documented API , because it is often convenient for bioinformatics to embed something in their code, so that we try to make the software as user-friendly as possible.
Now we come to how you can make a diagnosis from this data. Already, all these genome sequencing experiments are helping to come up with some new drugs. But to use this data as such, you need to learn how to extract patterns from it.
It is indeed possible, many articles show that the same CDR3 is produced for the same types of infections. This suggests that diagnosis is possible. It is clear that there are complex objects that give rise to not very pronounced patterns. And in order to detect such patterns, we need a large statistic, a large set of both control data (healthy donors), and sick patients with known diseases. Most likely, one clinic will not be able to accumulate such a number of samples in order to trace the necessary patterns for a large range of diseases. And a single place for collaboration will contribute to the development of diagnostics.
Such a database should store this data in some convenient structure that allowed them to make specific casts necessary for calculating patterns and analyzing samples. It should delimit access, because many clinical data can not be spread in public access. And it is also necessary for this database to do its job: to catch data patterns, and then look for them in new data, gradually update them through the same new data, improve its performance.
Such a base is set for a long-term perspective. From the very beginning it may not be clear why it is needed. But this data is complex in itself. Imagine that you are working with hundreds of arrays, each with a million sequences. In this case, you need to look for patterns between them (and they are quite complex), and you are not necessarily looking for fully equivalent CDR3, but, for example, similar ones. And the search is carried out immediately in many arrays. In general, it is very difficult to find patterns. We need some kind of high-level language that would allow all this to be described. Now researchers, who work with this data, have to write some scripts each time. In fact, to trace some clear patterns is smarter than just finding the same clones of the same arrays in the same arrays, now it just does not work out, there are no necessary tools. Therefore, if you make such a base, it will be interesting to people, because solve their specific research problems. And all this should be convenient and clear, since biologists often work with this data, for whom a graphical interface is simply necessary. We need a platform for collaboration, because for really interesting results you need a lot of data that would flock to one place from different sources.
The main problem in organizing such a common database is the delivery of this data from researchers. We are talking about hundreds of GB, and even terabytes. Gradually, this problem begins to be solved thanks to cloud services. Many researchers upload their work to Yandex.Disk, Dropbox, etc. There are also specialized cloud storage. For example, BaseSpace - sharpened specifically for Illumina sequencers. Those. When you buy a sequencer, you automatically get an account in BaseSpace, and the data is loaded into it directly during the sequencing process. In addition, BaseSpace has a convenient API through which external applications can access it. So it can be used as part of the infrastructure.
We already have some prototype database. The idea is that if the raw data potentially needs to be reprocessed after some time, then they can be written to tape and stored somewhere separate. This is the cheapest way to store large amounts of data. Such services are provided, for example, Amazon Glacier. Processed data can be stored in a relational database, but this is completely optional. In essence, this is unstructured data - CDR3 lists. Therefore, they can be stored as files in block storages and loaded them when rebuilding the database structure.
Bioinformatics, when calculating patterns at a low level, are likely to need the following queries:
To do this quickly and easily, not to write any proprietary algorithms, you can use PostgreSQL. Most likely, this is not the only way, but in the prototype we used it. In PostgreSQL, there is such a thing as a reverse key: in the fields you can just store arrays of ints, respectively, you can make queries like "give me all the records that contain the digits 3, 5 and 8 in these fields." And these requests are very fast. The picture below shows a simplified model of our prototype. There should also be a bunch of meta-information, other side information that comes with CDR3. In fact, we achieved good performance due to the fact that CDR3 served as keys, and the list of data sets in which this CDR3 participated, in which it met, was stored as data. Therefore, requests for this key are fully consistent with those requests that such a database will usually execute.
You can talk about the performance of the prototype in more detail. For example, let's take a machine with a quad-core Intel Core i7 2600 @ 3.8 GHz, 16 GB of RAM and software RAID1 from two SSDs of 128 GB each. 112 arrays were sent to the input, containing a total of 45 million colons. All this fit in 70 GB SSD. As a result, on requests like “all clones from two arrays” we managed to achieve a performance of 50 operations per second with a delay of about 150 milliseconds.
In conclusion, I would like to repeat that the accumulation of data in one place is necessary for such a thing to work as a diagnostic. Now all tests are aimed at determining whether a person has a particular disease. The new type of diagnostics is designed to give an answer with the help of one analysis at once, with what exactly the person is sick. To do this, you need to collect and process a lot of data, and our prototype shows that it is quite realistic and not very resource intensive.
Video of the report
')
We will start from afar so that it is clear what data we are working with and where they come from. The picture under the cut is very schematically depicted immunity. Cells that have the same specificity are colored with the same color (i.e., they recognize the same types of infections). We call these cells clones. During an infection attack, the number of cells that recognize it increases.
The specificity of these cells is due to the fact that they have a T-cell receptor on the surface, the assembly rules of which are recorded in the corresponding gene. For the subsequent narration, it is important to understand its structure.
A gene is a piece of DNA, you can think of it simply as a sequence consisting of a four-letter alphabet. The uniqueness of the T-cell receptor gene is that it is different in different cells. All other genes that are in our cells are the same, only the T cell receptor and antibody genes differ in T and B lymphocytes, respectively.
This gene consists of 4 main pieces. One piece in T-cell receptors is always the same. The other two sites (red and green in the picture) are selected from a small set. During the assembly of these two sections, random letters (about ten) are added between them, which leads to the formation of a giant variety of such receptors. The site that then recognizes an antigen in a protein is called CDR3.
It is interesting from two sides: on the one hand, it determines the specificity, because simply by biology it just encodes the receptor site responsible for the recognition of antigen, and on the other hand it is convenient as an identifier, because all diversity is concentrated in it, it includes a piece of red plot, a piece of green. From these pieces we can fully determine which of the set of these segments was chosen, and all these random letters are fully concentrated in it. Therefore, knowing CDR3 we can reconstruct the entire protein. We can also use it as an identifier, because if CDR3 is the same, then the whole gene is also the same.
So, we have a blood sample, we carry out some sequence of complex molecular biological reactions in order to isolate from it DNA sequences containing CDR3. Next, we load them onto the sequencer, and at the output we get something like that:
These are real data that come to us from the sequencing center. If you discard too much, then in fact it is just DNA sequences from 100 to 250 nucleotides in length. And there are a lot of such sequences.
Our task is to reconstruct from this data what was at the very beginning find out how many and which clones were in the primary sample.
Everything is complicated by the fact that in the data there are a variety of errors. These errors are introduced at all stages: while sample preparation takes place, while it is sequenced, etc. The rate of error can vary, depending on the architecture of the experiment from the sequencer that was used, in general, from everything. These can be inserts, deletions and replacements.
And this is bad, first of all, from the point of view that the data on the sample are distorted and artificial diversity is introduced. Suppose we had a sample consisting of only one clone, but as a result of the preparation, mistakes were made to it. As a result, we see that we have more clones than there were, and we have the wrong picture of the patient's T-cell repertoire.
Now briefly about what data we work with. At the entrance, we get about a hundred million sequences, or even a billion, 100-250 nucleotides in length. All this in total weighs up to 100 GB. And the concentration of CDR3 it can be from 1/5 (in very rare cases it is more) and up to the read sequences are millions.
The scheme of MiCTR software developed by us looks approximately as follows:
MiCTR solves the problem I described earlier: it makes a list of clones from the data and corrects errors. The first block extracts from the CDR3 data, the next blocks work only with CDR3 already and combine them into clones and correct the errors.
On the first block I will not concentrate much. He is engaged in leveling, and this is a huge part of bioinformatics, which would require a separate lecture. In short, this block lays out the input sequences, determining where the red is in them and where the green areas are. The set from which they are selected is known, it is in the databases and absolutely the same for all people. We determine which of these red areas is found and where. Do the same with green areas. Having built these alignments, we already know where we have CDR3, and we can select them from the sequences and work with them.
The most trivial thing to do with this data is to combine them by the same: the same CDR3 must be collected and counted. But since there are errors in the data that need to be corrected, and to correct them, we will subsequently perform a fuzzy search on multiple CDR3s (for example, search for CDR3 that differ by one letter), then for storage we use a prefix tree (trie), which is very convenient for such operations . In addition, when growing, this tree is not rebuilt, so it is very convenient to implement all competitive algorithms on it. No need to worry much about any tricky syncs. This tree is very easy to use, but it has its drawbacks. It is heavy and weighs quite a lot in RAM. However, in the real world, it turns out that this does not pose a particular problem (it can be kept within 1-2), so this is an ideal container for such data.
What do we do in relation to errors? In fact, we collect all these clones from only a part of the sequences. The fact is that the sequencer, in addition to giving the letters that it has read, puts in each position a value of quality that determines the probability of error. Those. The sequencer read the letter, but he can be sure of it or not. And for each position is set the probability of error. And here we are doing a fairly simple trick: we just carry out a certain error probability threshold, say, one-hundredth. All the letters that have a high probability of error, we call bad. Accordingly, all CDR3 containing such a letter are also considered bad. All this is stored in a container that not only collects data by number, but also collects other information: it searches for equivalent CDR3, or CDR3 that differ only in positions with poor quality. Thus it is possible to correct a large proportion of errors. Unfortunately, it is impossible to get rid of all errors, some errors are made before the sequencer, so there is no additional information about them.
And here we are doing a fairly simple thing, we find CDR3 pairs that differ by one letter and are very different in concentration. Most often, this is a clear marker for errors. And we simply remove them, choosing a threshold of the order of 1 to 10 by concentration: one error per sequence.
In sum, all this allows us to correct 95% of the errors and not to remove the natural diversity. It is clear that with such algorithms you can make a mistake and take the natural clone for an error, but in fact they are so diverse that this does not happen. Such performance is a good number for real work.
It is worth a little to describe the software implementation of all this. The system is multi-threaded, the sequence markup is launched in parallel. In uniprocessor systems, the best result is achieved if the number of threads matches the number of cores. The diagram above shows work in two streams: one is engaged in the formation of clones from good CDR3, the other from bad ones. All this scales well: for example, on a six-core processor, the execution of the code is accelerated approximately 3.5-4 times. As for performance, but on a completely ordinary hardware (AMD Phenom II X4 @ 3.2 GHz) 50,000 sequences per second are processed. The storage of one clone type takes about 5 KB of RAM. Thus, the most complex (diverse) array from our experience was able to process in 20 minutes on a machine with the above processor and 16 GB of RAM.
Everything is written in Java, the source is open , there is a well-documented API , because it is often convenient for bioinformatics to embed something in their code, so that we try to make the software as user-friendly as possible.
Now we come to how you can make a diagnosis from this data. Already, all these genome sequencing experiments are helping to come up with some new drugs. But to use this data as such, you need to learn how to extract patterns from it.
It is indeed possible, many articles show that the same CDR3 is produced for the same types of infections. This suggests that diagnosis is possible. It is clear that there are complex objects that give rise to not very pronounced patterns. And in order to detect such patterns, we need a large statistic, a large set of both control data (healthy donors), and sick patients with known diseases. Most likely, one clinic will not be able to accumulate such a number of samples in order to trace the necessary patterns for a large range of diseases. And a single place for collaboration will contribute to the development of diagnostics.
Such a database should store this data in some convenient structure that allowed them to make specific casts necessary for calculating patterns and analyzing samples. It should delimit access, because many clinical data can not be spread in public access. And it is also necessary for this database to do its job: to catch data patterns, and then look for them in new data, gradually update them through the same new data, improve its performance.
Such a base is set for a long-term perspective. From the very beginning it may not be clear why it is needed. But this data is complex in itself. Imagine that you are working with hundreds of arrays, each with a million sequences. In this case, you need to look for patterns between them (and they are quite complex), and you are not necessarily looking for fully equivalent CDR3, but, for example, similar ones. And the search is carried out immediately in many arrays. In general, it is very difficult to find patterns. We need some kind of high-level language that would allow all this to be described. Now researchers, who work with this data, have to write some scripts each time. In fact, to trace some clear patterns is smarter than just finding the same clones of the same arrays in the same arrays, now it just does not work out, there are no necessary tools. Therefore, if you make such a base, it will be interesting to people, because solve their specific research problems. And all this should be convenient and clear, since biologists often work with this data, for whom a graphical interface is simply necessary. We need a platform for collaboration, because for really interesting results you need a lot of data that would flock to one place from different sources.
The main problem in organizing such a common database is the delivery of this data from researchers. We are talking about hundreds of GB, and even terabytes. Gradually, this problem begins to be solved thanks to cloud services. Many researchers upload their work to Yandex.Disk, Dropbox, etc. There are also specialized cloud storage. For example, BaseSpace - sharpened specifically for Illumina sequencers. Those. When you buy a sequencer, you automatically get an account in BaseSpace, and the data is loaded into it directly during the sequencing process. In addition, BaseSpace has a convenient API through which external applications can access it. So it can be used as part of the infrastructure.
We already have some prototype database. The idea is that if the raw data potentially needs to be reprocessed after some time, then they can be written to tape and stored somewhere separate. This is the cheapest way to store large amounts of data. Such services are provided, for example, Amazon Glacier. Processed data can be stored in a relational database, but this is completely optional. In essence, this is unstructured data - CDR3 lists. Therefore, they can be stored as files in block storages and loaded them when rebuilding the database structure.
Bioinformatics, when calculating patterns at a low level, are likely to need the following queries:
- Find all the arrays that a particular clone meets.
- Find all the clones found in the array group.
- Various fuzzy searches and groupings
To do this quickly and easily, not to write any proprietary algorithms, you can use PostgreSQL. Most likely, this is not the only way, but in the prototype we used it. In PostgreSQL, there is such a thing as a reverse key: in the fields you can just store arrays of ints, respectively, you can make queries like "give me all the records that contain the digits 3, 5 and 8 in these fields." And these requests are very fast. The picture below shows a simplified model of our prototype. There should also be a bunch of meta-information, other side information that comes with CDR3. In fact, we achieved good performance due to the fact that CDR3 served as keys, and the list of data sets in which this CDR3 participated, in which it met, was stored as data. Therefore, requests for this key are fully consistent with those requests that such a database will usually execute.
You can talk about the performance of the prototype in more detail. For example, let's take a machine with a quad-core Intel Core i7 2600 @ 3.8 GHz, 16 GB of RAM and software RAID1 from two SSDs of 128 GB each. 112 arrays were sent to the input, containing a total of 45 million colons. All this fit in 70 GB SSD. As a result, on requests like “all clones from two arrays” we managed to achieve a performance of 50 operations per second with a delay of about 150 milliseconds.
In conclusion, I would like to repeat that the accumulation of data in one place is necessary for such a thing to work as a diagnostic. Now all tests are aimed at determining whether a person has a particular disease. The new type of diagnostics is designed to give an answer with the help of one analysis at once, with what exactly the person is sick. To do this, you need to collect and process a lot of data, and our prototype shows that it is quite realistic and not very resource intensive.
Source: https://habr.com/ru/post/228003/
All Articles