How to create a 3D-model of a human virus. Part One: Collection and Analysis of Scientific Data

At the end of the movie Blade Runner, Rutger Hauer's hero says that he has seen a lot that people can't even imagine - space battles, flaming warships - but is it so difficult to imagine all this for a modern person spoiled by computer graphics? At the same time, we are surrounded by many things that we can never consider in detail, due to fundamental physical limitations. Moreover, we ourselves consist of such things. However, the good news is that these objects can be modeled and visualized using the currently developed 3D toolkit. And if you assemble a team in which there will be not only three-dimensional modelers, visualizers and designers, but also scientists, you can bring the result as close as possible to reality.
Under the cut, the first part of the story about our experience in creating scientifically reliable models of viruses.
The world of molecular machines and viruses offers a lot of interesting challenges for CG teams. The problem is that so far there is no universal scientific methodology that would allow to fully describe the structure of the virus particle. In order to describe the device of the virus you need to use a variety of methods that give an idea of the individual pieces of the final puzzle. Electron microscopy makes it possible to estimate the size and shape of virions, X-ray analysis can describe individual proteins or their fragments, and molecular-biological and biochemical methods provide information about how many molecules are included in the virus and how they interact with each other. This creates a somewhat paradoxical situation: many viruses are studied in great detail and in detail, but there are no images that would give a scientifically reliable and complete picture of how they are arranged.
')
For example, modern electron micrographs of viral flu particles look like this ( source ).
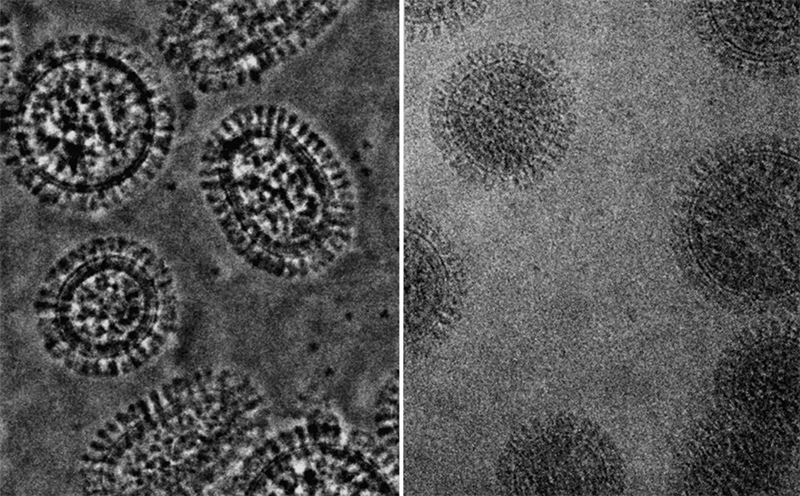
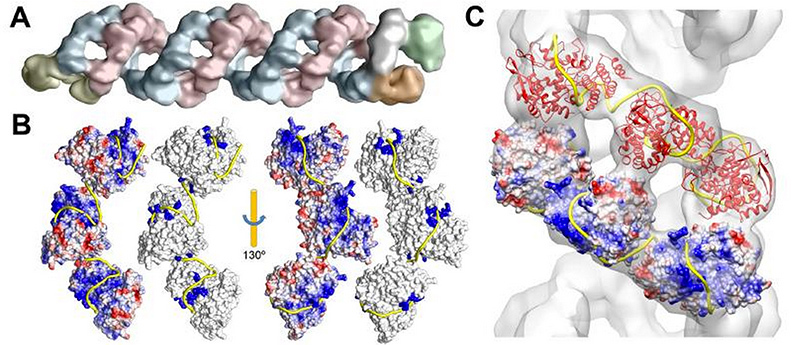
Data visualization of cryoelectron microscopy of the genomic complex of influenza A virus and reconstruction of the RNA (yellow ribbon) package with proteins B and C. A group of virologists from Madrid, who helped us create the A / H1N1 influenza virus model, published this data in the journal Science Science in late 2012.
To collect all available information is technically possible. But its systematization, processing and translation into the 3D model requires a team approach. At the same time, even a competent scientific consultant cannot have a full baggage of highly specialized knowledge on the topic, so it is important to connect scientists who have dedicated their entire career to working with a virus. A modeller without biological education will not understand the published scientific data and protein structures from the Protein Data Bank , and will not be able to correctly complete the molecular model using molecular dynamics, where necessary (approximately 80-90% of the proteins we encounter have an incomplete description spatial structure by 10-90%). A scientist, even having all the information separately, cannot collect and visualize the full model in professional programs for three-dimensional modeling. In our experience, only the close interaction of these specialists can give a neat and informative result.
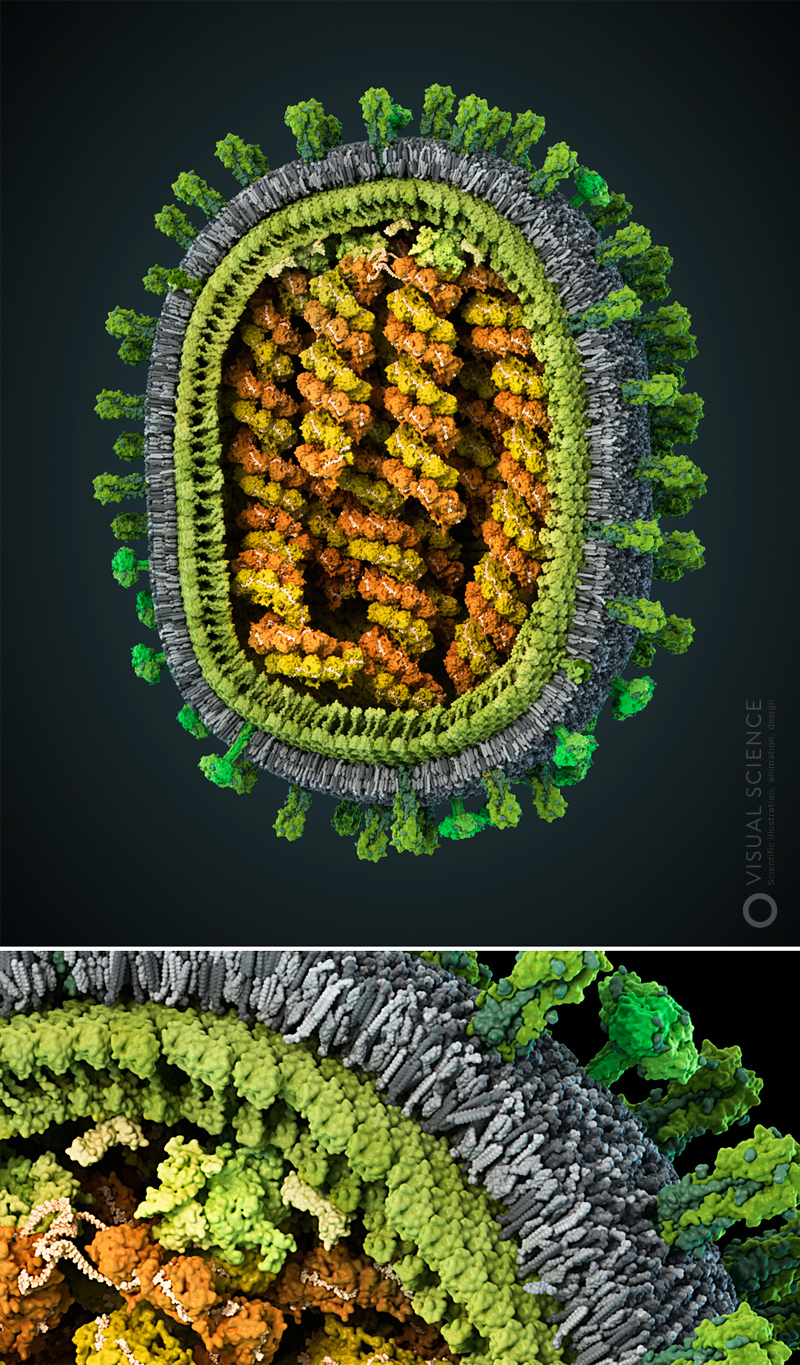
Image of influenza virus with the detail to the atoms. All proteins and protein complexes in the composition of the particle, as well as their quantitative ratios and position correspond to the data published in the scientific literature ( signatures of all components ). The model was created with the participation of Jaime Martin-Benito and colleagues (Spanish National Center for Biotechnology, Madrid, Spain). year 2013.
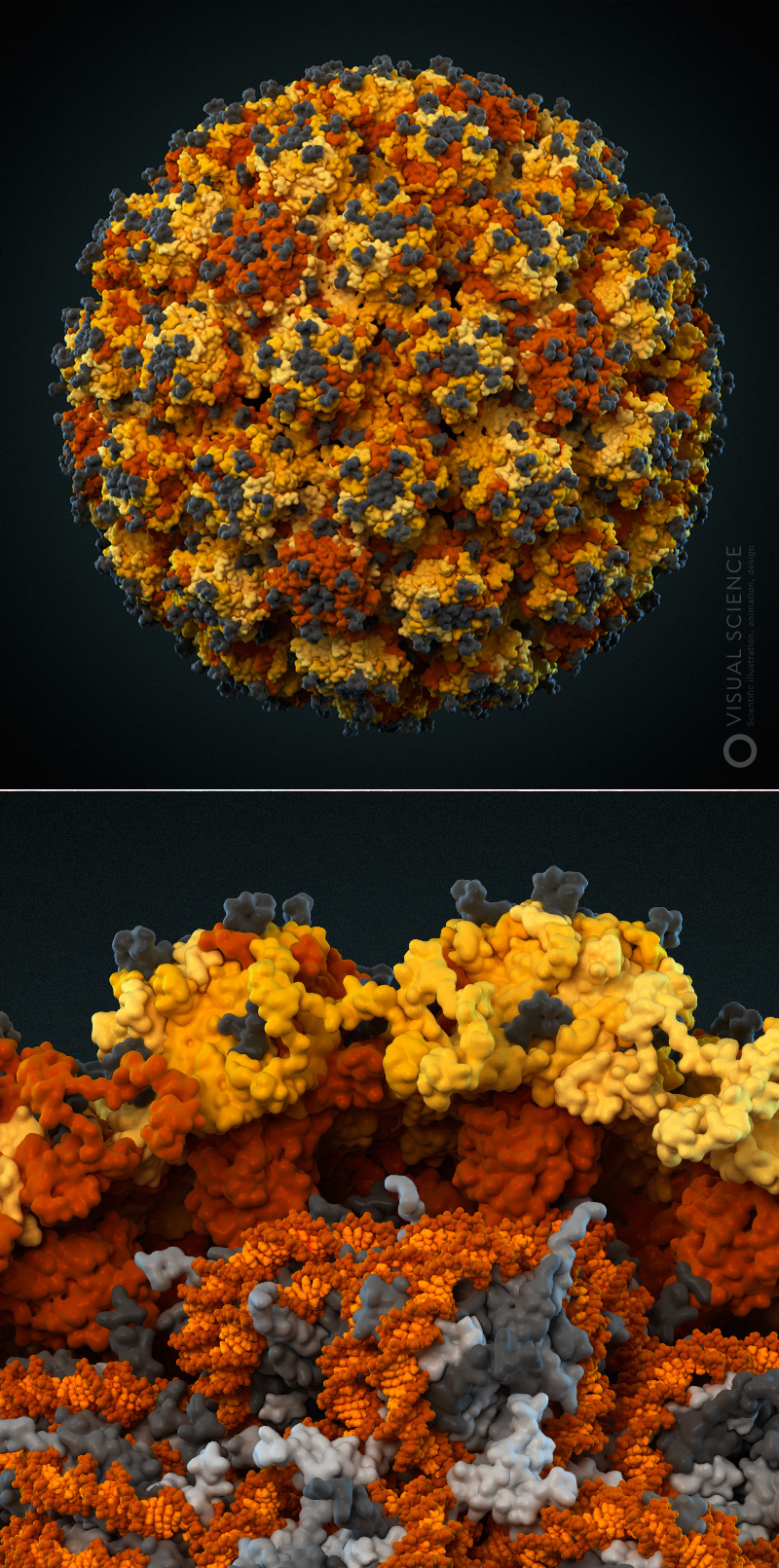
Internal structure of human immunodeficiency virus. The edge of the membrane membrane, proteins present inside the virion, the capsid and fragments of the virus RNA, enclosed in it (the signatures of all components ) are visible. The model was created with the participation of Egor Voronin (Global HIV Vaccine Enterprise). Prize for the best scientific illustration at the Science and Engineering Visualization Challenge in 2011.

Model of the alleged folding of the human papillomavirus genome. The model was created with the participation of Christopher Buck (National Cancer Institute, USA). year 2012.
Particle and individual proteins of the Ebola virus. The model was created with the participation of Ronald Harty (University of Pennyslvania, USA). Honorable mention of the Science and Engineering Visualization Challenge in 2010. Exposition of the Salon of the Association of Medical Illustrators in Toronto in 2012.
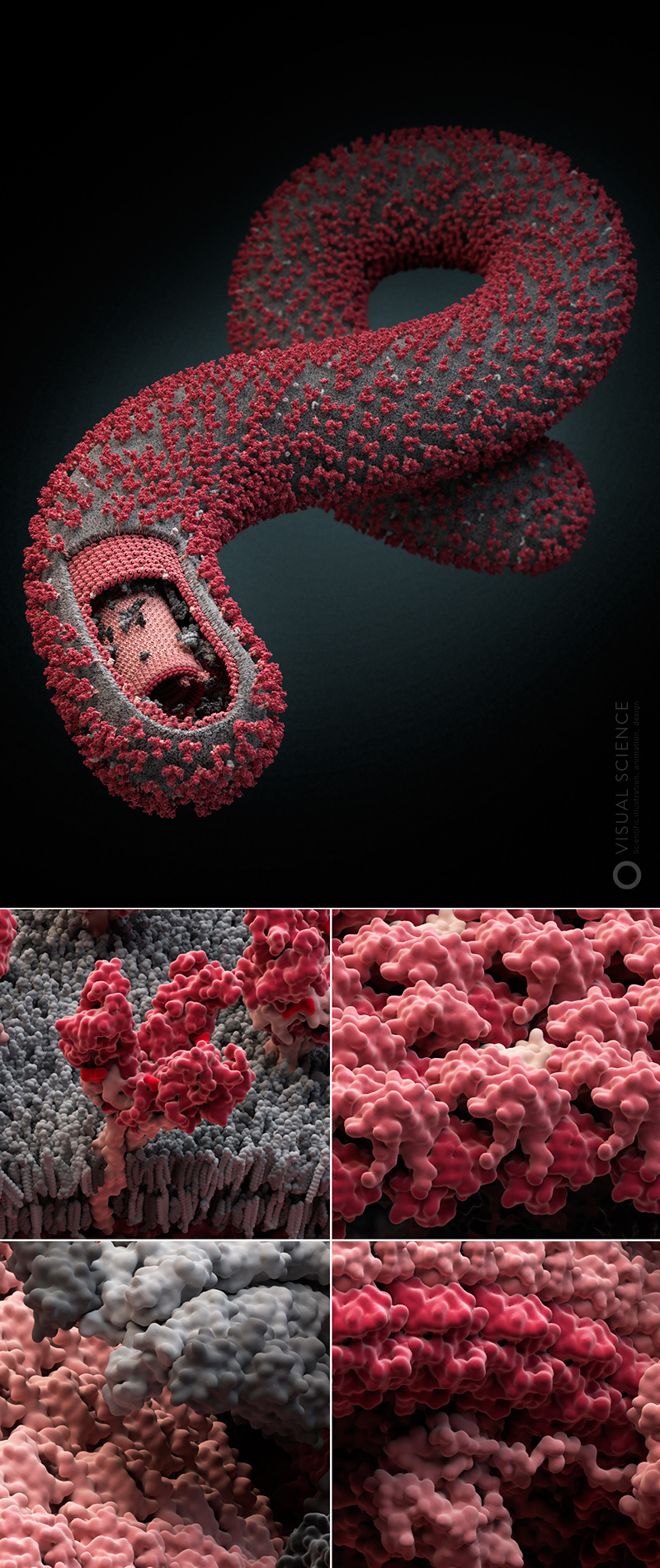
Our studio several years ago launched a non-commercial project, the essence of which is in modeling and visualizing the most common and dangerous human viruses. We called it Viral Park, or “Virus Zoo”. The project still includes four viral models, a few more are in development, and plans to make a series of about twenty virions. During the work on the project we managed to master and adjust the process, highlighting in it a number of stages:
- Literature review and systematization of detected data
- Molecular modeling and dynamics
- Build a complete model from individual elements
- 3D visualization and design
- Creating materials based on the model from posters to applications, widgets and plastic models.
In this post we will talk a little about the first stage of our work.
Gathering information about the topic being studied is a task that scientists solve all the time. It is impossible to make a new project without knowing what was published before you. For this, it is necessary to find and analyze first review and then research publications on the subject of interest. The same scheme works when information about the structure of viruses is collected. Thanks to the databases of natural-science publications of the world's leading journals PubMed and Google Scholar, this process can be organized very effectively. If you need background information about the biology of the virus, you can use the Viral Zone site and a lot of data on individual proteins is available in the Uniprot database. The structures of proteins or their fragments, obtained by different teams of scientists using the methods of nuclear magnetic resonance and X-ray analysis, are available in the already mentioned Protein Data Bank in the form of coordinates of all atoms or, in some cases, only alpha atoms of the protein chain.
The task for a scientist in the process of creating a virus model is to collect, process and prepare all the information in a form that will be convenient for the work of the rest of the team. It is necessary to make a complete list of all types of molecules that form a particle, and all their interactions. In addition to proteins, these can be membrane lipids and viral genome molecules represented by DNA or RNA. Next you need to understand in what quantities the molecules are represented in the particle, and what places they occupy. This is the most difficult to find and often contradictory and incomplete information, since different methods may give different estimates. To clarify certain issues, we contact the authors of the articles in which they are discussed. This is a well-accepted practice in the scientific community, and scientists are often happy, and sometimes without contact, and sometimes share their hypotheses and even unpublished data, as was the case when working on the Influenza model in the case of the already mentioned Spanish virologists.
The result of the study of the literature should be the most detailed verbal picture of the future model. It is necessary to understand what, in what quantities and in what way is packaged in a virus particle. This can be summarized in a description, a table of quantities and interactions, and a plan of the model at the desired scale.
Further stages of work imply obtaining three-dimensional models of all necessary components. One of the problems here is that atomic structures may not be available for all proteins and their complexes. Scientists have not yet been able to describe a substantial part of viral proteins. In our work, we use structural bioinformatics methods to fill this gap. We will tell about it in the following posts. We will also try to reveal the details of how the full model is assembled, its visualization and the creation of educational tools and widgets based on the result obtained.
We believe that such a detailed approach to the modeling of molecular biological objects has great promise in terms of its application in education, popularization of science and scientific communication. This is supported by the fact that such models receive high marks at major international competitions of scientific illustration and design, positive reviews of well-known colleagues, and it is pleasant to include such images in their presentations even by Françoise Barré-Sinoussi , who won the Nobel Prize for discovering HIV.
In continuation of the topic, in addition to modeling viruses in the framework of the Zoo of viruses, we will discuss the scope of scientific and medical illustration in general, let's talk about why this is relevant, how it differs from popular science, or Science Art, and how it will help make the world a better place. and science is clearer.
Source: https://habr.com/ru/post/222819/
All Articles