Again on bioinformatics: the assembly of bacterial genomes
 Since I published the first review article on bioinformatics , almost a year has passed. I would like to write more often, but just bioinformatics is distracted and does not give. In the previous article, the task of assembling the genome was mentioned, as well as the Laboratory of Algorithmic Biology ( SPbAU RAS ), in which it is engaged. As already mentioned, this task has been worked in many universities of the world for a long time and quite successfully. However, to assemble a genome, biologists get a huge amount of different types of data, each of which has its own characteristics. About five years ago, MDA technology appeared, which opened up great opportunities in the field of studying bacteria. As a result, a data type emerged that required a new genomic collector. A few years later it was developed, and not at Stanford or Massachusetts, but in St. Petersburg, at the young Academic University. But first things first.
Since I published the first review article on bioinformatics , almost a year has passed. I would like to write more often, but just bioinformatics is distracted and does not give. In the previous article, the task of assembling the genome was mentioned, as well as the Laboratory of Algorithmic Biology ( SPbAU RAS ), in which it is engaged. As already mentioned, this task has been worked in many universities of the world for a long time and quite successfully. However, to assemble a genome, biologists get a huge amount of different types of data, each of which has its own characteristics. About five years ago, MDA technology appeared, which opened up great opportunities in the field of studying bacteria. As a result, a data type emerged that required a new genomic collector. A few years later it was developed, and not at Stanford or Massachusetts, but in St. Petersburg, at the young Academic University. But first things first.Again about the genome assembly
Briefly recall basic concepts. The genome for biologists is a long DNA molecule, which is a double chain of four types of nucleotides (adenine, cytosine, guanine and thymine). For example, the length of a DNA chain in a bacterium is measured in millions of nucleotides, while the length of human DNA is approximately 3 billion. For bioinformatists, the genome is just a big line above the alphabet of four characters {A, C, G, T}. “Read” the entire DNA molecule is completely impossible. Modern technology allows you to read only small pieces of a length of several hundred nucleotides from random places, and then with errors. These pieces are called reads or reads, and the reading process is called sequencing. In one experiment, several tens or even hundreds of millions of readings are performed. Next comes the bioinformatics that develop genomic assemblers - programs that attempt to restore the original sequence from these readings. As a rule, assemblers do not manage to restore the entire genome as a whole, but from millions of short readings they allow to get dozens of sequences hundreds of thousands of nucleotides in length. Such sequences can already be further studied by biologists.
 Who may need to collect and analyze the genome? For example, the assembly of the human genome can help determine the presence of cancer cells in the body at an early stage. Another goal - the transition to the so-called personal medicine. If each person has a known genome, then medications and treatment can be prescribed to him not only by symptoms, but also taking into account his genetic characteristics. Personal medicine is not yet widespread, but some clinics already provide patient sequencing services. The price of reading the genome is constantly falling, and new assembly algorithms allow it to be faster. However, the human genome alone is not enough for a complete diagnosis. In each of us there are several thousand species of different bacteria that are involved in a huge number of internal processes and are necessary for life. The assembly of the genomes of these bacteria (the total length of which exceeds the length of the human genome itself) would allow a much better study of the human body. However, there are difficulties in their sequencing.
Who may need to collect and analyze the genome? For example, the assembly of the human genome can help determine the presence of cancer cells in the body at an early stage. Another goal - the transition to the so-called personal medicine. If each person has a known genome, then medications and treatment can be prescribed to him not only by symptoms, but also taking into account his genetic characteristics. Personal medicine is not yet widespread, but some clinics already provide patient sequencing services. The price of reading the genome is constantly falling, and new assembly algorithms allow it to be faster. However, the human genome alone is not enough for a complete diagnosis. In each of us there are several thousand species of different bacteria that are involved in a huge number of internal processes and are necessary for life. The assembly of the genomes of these bacteria (the total length of which exceeds the length of the human genome itself) would allow a much better study of the human body. However, there are difficulties in their sequencing.From person to bacteria
To sequence a genome, it is necessary to have a sufficiently large amount of genetic material — identical DNA molecules. A standard experiment requires several tens or hundreds of millions of identical cells. In the case of multicellular organisms, including humans, this is not a problem, since a sufficient amount of genetic material is contained in a few grams of saliva. Obtaining as many bacterial cells is more difficult - they need to be grown in the laboratory. The problem is that not all bacteria can be cultivated. For example, bacteria that are inside the human intestines live in large colonies and are not able to exist and multiply separately from each other. It is impossible to cultivate a single bacteria taken from the colony.
')
Today, there are two fundamentally different approaches to the sequencing of uncultivated bacteria in colonies. The first, fairly simple from a biotechnological point of view, is metagenomics. It consists in the following: we take a colony, we select DNA molecules from all bacteria, we place them in a sequencer and we read. As a result, we will simultaneously receive the reading of the genomes of several dozen different bacteria - from some more, from some less. In closely related bacteria, the genomes are similar to each other, and in bacteria of different types, they are very different, but may contain identical areas. These factors greatly complicate the assembly of such data. Despite a number of metagenomic studies conducted and carried out today, I cannot say that there is at least one assembler that would give results on such data that would satisfy the biologists. As a rule, collectors give short sequences that contain a large number of errors and are difficult to analyze. Sometimes in metagenomic projects, the discovery of several genes for the entire set of different bacteria is already a success.
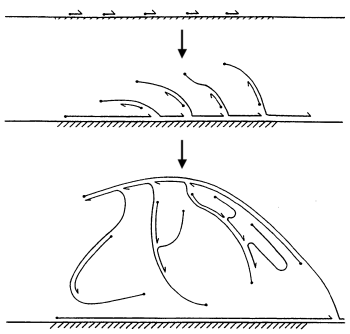 Another approach to sequencing uncultivated bacteria was discovered only a few years ago and consists of a rather complex biotechnological process. We take one bacterium of interest to us from the colony, extract DNA from it, and plant several “copying machines” on the DNA, which begin to “run” along the molecule and copy its parts randomly. When copying, new DNA strands branch off from the molecule, which in turn can also be joined by a “copy machine” and continue building up genetic material. As a result, we get identical copies of the sections of DNA of interest to us, which we place in the sequencer. This method of sequencing a single cell genome is called Multiple Displacement Amplification (MDA). It would seem that everything is great. But, alas, there are problems. “Copy machines” work on their own and cannot be controlled. Therefore, some sections of DNA can be copied ten thousand times, and others - never. As a result, some regions will be missing in the assembly simply due to lack of material. On average, as a result of this method, 95-99% of the genome can be restored, depending on its structure (some can be copied worse) and the quality of the experiment.
Another approach to sequencing uncultivated bacteria was discovered only a few years ago and consists of a rather complex biotechnological process. We take one bacterium of interest to us from the colony, extract DNA from it, and plant several “copying machines” on the DNA, which begin to “run” along the molecule and copy its parts randomly. When copying, new DNA strands branch off from the molecule, which in turn can also be joined by a “copy machine” and continue building up genetic material. As a result, we get identical copies of the sections of DNA of interest to us, which we place in the sequencer. This method of sequencing a single cell genome is called Multiple Displacement Amplification (MDA). It would seem that everything is great. But, alas, there are problems. “Copy machines” work on their own and cannot be controlled. Therefore, some sections of DNA can be copied ten thousand times, and others - never. As a result, some regions will be missing in the assembly simply due to lack of material. On average, as a result of this method, 95-99% of the genome can be restored, depending on its structure (some can be copied worse) and the quality of the experiment.MDA technology issues
The number of times that the nucleotide in the genome is read during the sequencing process is called its coating. In standard sequencing, the coverage of all nucleotides is approximately the same (an example of the coverage graph in the left figure below). When using the MDA technology, we obtain data with extremely uneven coverage (the right figure below), which is a critical problem for the assembly task. Let's try to figure out why.
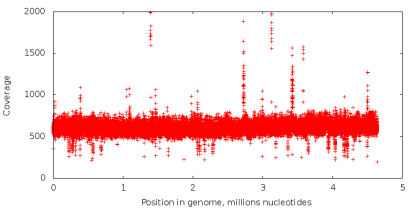
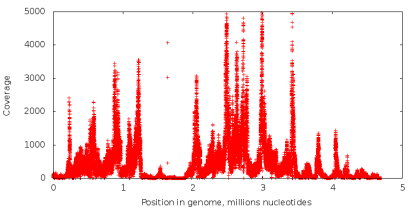
Let's make the following experiment. Take the data obtained using standard sequencing, and from all readings we extract substrings of a fixed length k (next - k-measures), so that from each possible position in reading one k-measure begins (in this way the ACGTAC sequence is divided into four 3-measures : ACG, CGT, GTA and TAC). Next, let's calculate how many times each k-mer occurs in our readings. If some k-mer appears a sufficiently large number of times (that is, it has a high coverage), there is reason to assume that it also occurs in the original genome, which we are sequencing. If k-mer is rare, then it most likely contains an incorrectly read nucleotide. Sequencing errors are a random event, and the likelihood that the same error will occur immediately in a large number of k-measures is very small (the probability of incorrect reading of a single nucleotide ranges from 0.001% to 1% depending on the position of the nucleotide in the reading). Thus, with the help of the value of the coverage of k-mers, it is possible to divide them into reliable and erroneous. Similar methods are used in most modern assemblers and utilities for correcting errors in readings. However, in the case of sequencing a single cell, it is impossible to divide k-measures in this way, since k-measures that are rarely encountered may correspond to a section copied a small number of times, and do not contain errors at all.
Uneven coverage is not the only problem with the MDA approach. When copying DNA from the main thread, a new single-stranded thread branches off. It can randomly stick together with another spun off thread, which is why DNA sequences will be next to it, which correspond to completely different regions in the original genome. In the place of such an erroneous gluing together, “copying machines” can again sit down and multiply the wrong part. Then, as a result of sequencing, we obtain readings containing sequences from different parts of the genome (chimerical readings). When processing such readings, the genomic collector may incorrectly combine two sequences into one. If one wrong nucleotide is not a gross error, then two wrongly glued sections are a much more serious problem that complicates further analysis.
At last
It is the data obtained using the MDA technology that became the main subject of research at the Laboratory of Algorithmic Biology in St. Petersburg. Many algorithms have been developed that have allowed to circumvent the problems of uneven coverage and chimerical readings. These algorithms were implemented in the genome assembler SPAdes, which today is the leader in the field of bacterial genome assembly and is used by leading global laboratories (for example, in the United Genomic Institute in the USA - the world's largest center for the study of bacterial genomes).
Then I would gladly acquaint readers with the basic principles of modern standard assemblers, talk about new approaches and algorithms that are used in the collection of data obtained in a single cell, but, unfortunately, this does not fit into one article. For those who are interested to learn more about the assembly algorithms - below there are links to popular texts and scientific publications on this topic. I sincerely hope that the next such article will appear faster than this time.
For those interested
- The function of the “copy machine” is performed by Phi-29 DNA polymerase, a protein capable of replicating DNA segments in random places.
- In order to identify and eliminate sequencing errors in data obtained using MDA, approaches based on, for example, clustering of k-mers by Hamming distance, topological features of the de Bruyne graph in places of errors are used.
- Search and removal of chimerical compounds are also mainly performed according to the topology of the graph de Bruijn.
Bibliography
- PEC Compeau and PA Pevzner. Genome Reconstruction: A Puzzle with a Billion Pieces (review article on genome assembly)
- PA Pevzner, H. Tang, and M. Waterman. An Eulerian path approach to DNA fragment assembly. Proceedings of the United States of America, 98: 9748-9753, 2001 (first article on the use of Count de Bruin in the task of assembling the genome)
- D. Zerbino, and E. Birney. Velvet: Briefs of the Bruijn graphs. Genome Research. 18 (2008), 821–829 (classic Velvet assembler)
- A. Bankevich, S. Nurk, et al. SPAdes: A New Genome Assembly Algorithm and Single-Cell Sequencing. Journal of Computational Biology 19 (5) (2012), 455-477 (SPAdes - assembler for assembling data obtained using MDA)
Source: https://habr.com/ru/post/186492/
All Articles