Support vector machine for finding polymorphisms in the genome
 The 2013 article “A support vector for single-nucleotide polymorphisms from next-generation sequencing data” (O'Fallon, Wooderchak-Donahue, Crockett) proposes a new method for determining polymorphisms in the genome using the support vector machine (SVM ). Although earlier in the 2011 article, “The framework for variation discovery and genotyping using next-generation DNA sequencing data,” has already described the use of machine learning methods for determining single-nucleotide polymorphisms ( SNPs , snaps), an approach based on the use of SVM, is described for the first time in this article.
The 2013 article “A support vector for single-nucleotide polymorphisms from next-generation sequencing data” (O'Fallon, Wooderchak-Donahue, Crockett) proposes a new method for determining polymorphisms in the genome using the support vector machine (SVM ). Although earlier in the 2011 article, “The framework for variation discovery and genotyping using next-generation DNA sequencing data,” has already described the use of machine learning methods for determining single-nucleotide polymorphisms ( SNPs , snaps), an approach based on the use of SVM, is described for the first time in this article.Determining polymorphisms in the genome is important (for example, for a full genomic search for aka GWAS associations ), but not a trivial task. It is necessary to take into account that many organisms are heterozygous, and also that the data may contain erroneous information.
The standard approach to the determination of SNP is based on the alignment of sequencing data (reads, genome fragments) relative to the reference (reference) genome. However, reads may contain errors and may be incorrectly aligned to the reference. Since it is believed that the occurrence of an error in this position is a rarer event than the appearance of polymorphism, the simplest approach in this case would be to filter out the options with a small frequency. However, in the 2010 article, “Uncovering the whole genome sequencing” , it was shown that rare polymorphisms that occur with a frequency of <1% have the greatest effect on many diseases. Frequency-based filtering will entail a large number of false negatives for rare SNPs, so there is a need to create more sensitive methods for determining genomic variations.
As stated in the article, the advantage of methods based on machine learning is that they allow you to combine various factors affecting the plausibility of the occurrence of polymorphisms in a given position in the genome, which, among other things, increases the sensitivity of the method to rarer polymorphisms.
')
Unlike other machine learning methods (for example, the reinforcement learning approach described in the above article ), SVM requires significantly less data to train.
This article does not describe the detailed method for implementing the algorithm, but provides links to other articles containing technical details. LIBSVM is used as a platform providing an implementation of the SVM algorithm. The first publication about LIBSVM refers to 2011, and the latest release on the site to 2013. It should be noted that SAM or BAM files are accepted as input data, which seems to be a convenient and natural approach to bioinformatics. As a result of the algorithm, a VCF file is generated.
Two types of data are used to train the model:
- alignments corresponding to true polymorphisms;
- nucleotide mismatches in certain positions not related to the presence of polymorphisms.
I wonder how you can collect data corresponding to the second type. The authors cite as an example two possible strategies:
- Identify polymorphisms using GATK's best practices guidelines , including UnifiedGenotyper . The authors used snaps, which were determined only in one case (while excluding snipes, which are described in the data of the 1000 Genomes project), from the remaining ones, snaps were chosen with a very low leveling quality value.
- The authors chose polymorphisms that occur at least 8 times in the local database of 57 exomes (which also do not correspond to data from 1000 Genomes).
It should be noted that polymorphisms can also occur in non-transcribed regions of the genome; therefore, it is possible that the use of information obtained solely from the analysis of exomes may entail some inaccuracies.
On the received data training is performed. The article states that the training includes the determination of the numerical parameters of the hyperplane that best separates the two classes of data on which the training is performed.
It is also interesting, what signs were chosen by the authors for training the model. The authors taught the model, starting with a set consisting of three signs. This gave a bad result, so many of the signs expanded. This process is described in the article not very detailed, and it is not entirely clear in which order the authors added new signs, because it is obvious that they did not go through all the existing subsets of features. In total, the authors considered 16 features, among them the probability of an error in the reads, the average quality of a given nucleotide in the reads, the sum of the qualities of the nucleotides in a given position (the last two did not give any improvement when added to the set of features), the quality of the read alignment, the depth of sequencing also did not improve the work of the model), the balance of alleles, the quality of nearby nucleotides.
According to the graph provided in the article, the described method is superior to other methods for determining polymorphisms in sensitivity and specificity on a particular NA12878 dataset . The method identified 95.6% of all true positives, while other methods: UnifiedGenotyper, UnifiedGenotyper + VQSR , SAMtools and FreeBayes identified only 90.6%, 94.1%, 89.5% and 88.7%, respectively.
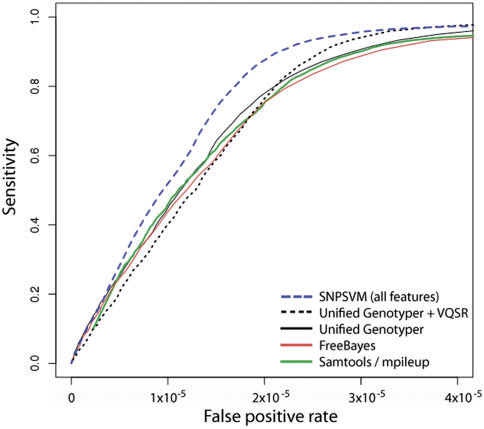
Among the shortcomings of the described approach, it can be noted that the SVM model can give poor results on data containing different error profiles. In addition, despite the multithreading, the implementation was 10-20% slower than the GATK UnifiedGenotyper method.
In general, the chosen model fits well with the specifics of the task and corresponds to the general trend of using machine learning methods for analyzing genomic sequences.
The article was written as part of a bioinformatics workshop .
Source: https://habr.com/ru/post/183330/
All Articles