Protein Structure: An Introduction for IT Professionals
It is nice to see that habravchane regularly interested in other subject areas - for example, biology (more specifically, the structure and function of biological macromolecules). However, some posts (for example, this one ) cause a specialist simply physical pain due to the abundance of completely wild factual errors. In this post I want to talk about the structure and function of the protein. About what we know and about what we don’t know, as well as about the computational tasks in this area that require solutions and are interesting for IT specialists. I will try to talk concisely and tezisno, to have more information, and less water. Anyone interested in the structure of proteins, please under the cat, there are a lot of letters.
As Friedrich Engels said, “Life is a way of existence of protein bodies”. In the 19th century, they still did not know about the role of DNA in the inheritance of genetic information, but Uncle Frederick’s statement is still largely true - proteins do most of the work in our cells. This includes maintaining the structure (cell shape), chemical catalysis, and motor function (muscle contraction, for example), and transport (say, hemoglobin protein transports oxygen from the lungs to the tissue and carbon dioxide in the opposite direction) and complex regulatory functions to maintain consistency internal environment (for example, protein hormones and all intracellular regulatory systems) and many others. In a word, if something happens in our body, proteins are necessarily involved (though not only them).
From a chemical point of view, a protein is a linear (unbranched) polymer consisting of monotonously repeating identical blocks of the “main chain”, to which various “side groups” are attached. Since the main chain blocks are asymmetric, the entire polypeptide chain of the protein has a direction, the N- and C-termini of the polypeptide chain are distinguished.
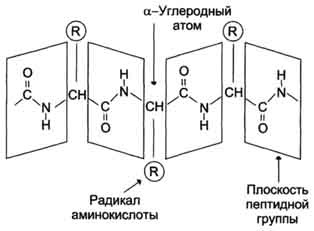
The chain length is from 70 to more than 1000 monomers (amino acid residues), the average length for higher organisms is about 500-600 amino acid residues, for bacteria this value will be less, most likely 300-400 residues. In total, there are 20 standard amino acids in nature, the same for both bacteria and humans, that is, 20 different side groups can stick out from the main chain.
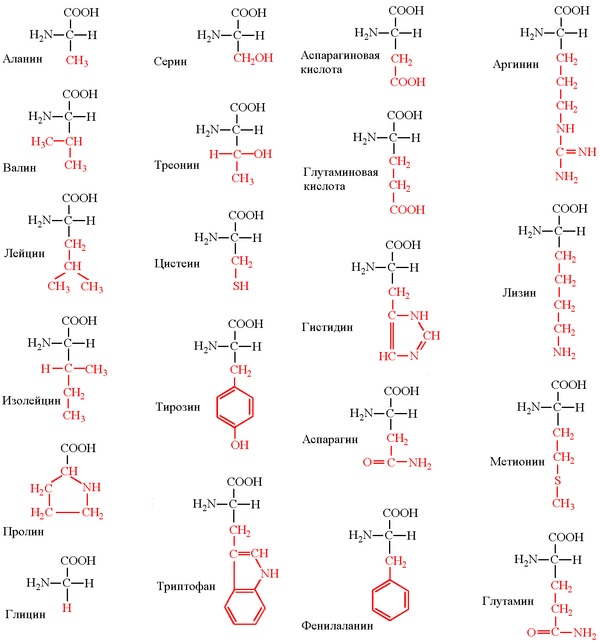
(A correction is possible here - some chemical groups can be modified after protein synthesis, for example, phosphorylated. However, this is not considered as another amino acid, but is considered as a product of the original modification. Also, higher non-canonical amino acids are possible in higher organisms, but this is a rare event. That is, strictly speaking, 22 different amino acids, 20 of them are basic and 2 rare, plus some side groups can be rarely chemically modified).
From generation to generation, genetic information is transmitted in the form of DNA, there are so-called “protein-coding regions” in it. In these places, DNA unambiguously (for botans - up to alternative splicing and RNA editing) encoded information about the linear amino acid sequence for the synthesis of this protein, plus there are corresponding machines in the cell that can synthesize a protein according to information originally encoded in DNA.
')
Since protein is a linear polymer assembled from 20 standard monomers, its so-called “primary structure” can be easily represented as a string, for example:
This is the amino acid sequence of a small human protein in the FASTA format, the first line starting with “>” describes its name, followed by the amino acid sequence in accordance with the standard coding (for example, M is methionium, S is serine, etc., a total of 20 letters of standard single-letter code), on the left is the N-terminus of the protein, on the right is its C-terminus. For different proteins, the length of the string will be obviously different, since proteins have different lengths. The sequences of all known proteins can be found in the public domain here: www.ncbi.nlm.nih.gov
Well, the primary structure is sorted out, but does the protein work in an expanded linear form? Of course not. Here it should be noted that from a structural point of view there are different classes of proteins: globular, membrane and fibrillar. Membrane proteins, as the name implies, live only in cell membranes, to stabilize their structure, you need a special membrane environment, we will not consider them in this review. Fibrillar proteins have a simple regular structure, look like elongated fibers, they are insoluble in water and perform structural functions (for example, hair consists of keratin, fibrillar proteins include protein from natural silk). Recently, they began to distinguish a class of disordered proteins — proteins that do not have a constant three-dimensional structure, or that acquire it only for a short time when interacting with other proteins. The most interesting from a practical point of view, the class of proteins, which we will consider - globular water-soluble proteins, most proteins belong to this class.
A linear polypeptide chain in water is capable of spontaneously folding into a complex three-dimensional structure (globule) and only in such a folded form can proteins perform chemical catalysis and other interesting work. Therefore, it is fundamentally important for us to know exactly the three-dimensional folding of the protein, since it is only at this level that it becomes clear how the protein works.
Question : how many three-dimensional structures correspond to a specific protein?
Answer : One, up to a small mobility of small “disordered” loops. Only one exception is known, when two sufficiently different structures correspond to one sequence, these are prions .
Question : Why does the protein have only one three-dimensional structure?
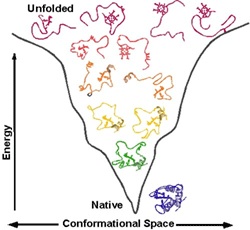
Answer : for chemical catalysis, we need to arrange the corresponding chemical groups in a strictly defined way in space. This requires a rigid structure. That is, the whole protein must be rigid in order to maintain the chemical groups of amino acids of the active center in the right places (in reality, many proteins consist of two or more rigid parts that can move relative to each other, this is necessary for regulating the activity of the protein ( allosteric regulation ) so that a certain signal could turn on and off the chemical activity of the protein-enzyme). To make the structure rigid and stable, nature made sure that the structure of each protein corresponded to the energy minimum of this system of atoms and this minimum was so deep that the protein did not “jump out” from it. All other parasitic structures have more energy and the protein still falls into the energy minimum corresponding to the native structure.
Question : what keeps the three-dimensional structure of the protein?
Answer : In short, mainly on a large number of non-covalent interactions. In principle, chemical groups of a protein can form: (1) a hydrogen bond, these groups exist in the main chain and in some side groups, (2) an ionic bond is an electrostatic interaction between oppositely charged side groups, (3) Van der Waalsovo interaction and (4) the hydrophobic effect, which keeps the overall structure of the protein. The point is that there are always hydrophobic aromatic residues in a protein, it is energetically unprofitable for them to contact with polar water molecules, and it is advantageous to “stick together” with each other. Thus, when the protein is folded, the hydrophobic groups are pushed out of the water environment, “sticking together” with each other and forming a “hydrophobic core”, while the polar and charged groups, on the contrary, tend to the water environment, forming the surface of the protein globule. Also (5), the side groups of two cysteine residues can form a disulfide bridge between them — a full-fledged covalent bond that rigidly fixes the protein.
Accordingly, all amino acids are divided into hydrophobic, polar (hydrophilic), positively and negatively charged. Plus cysteines capable of forming a covalent bond between each other. Glycine has special properties - it does not have a side group that severely restricts the conformational mobility of other residues, so it can “bend” very much and is located in places where the protein chain must be unfolded. In Proline, on the contrary, the side group forms a ring covalently linked to the main chain, rigidly fixing its conformation. Prolines are found where it is necessary to make the protein chain rigid and stiff. Many diseases are associated with the mutation of proline to glycine, due to which the structure of the protein slightly “floats”.
Question : how do we even know about the three-dimensional structures of the protein?
Answer : from the experiment, this is absolutely reliable data.
Now there are 3 methods for experimentally determining the structure of a protein: nuclear magnetic resonance (NMR), cryo-EM (electron microscopy), and x-ray diffraction analysis of protein crystals.
NMR allows you to determine the structure of the protein in solution, but it works only for very small proteins (for large ones it is impossible to make deconvolution).

This method was important for general evidence that a protein has only one three-dimensional structure and that the structure of a protein in a crystal is identical to the structure in solution. This is a very expensive method, since it is required to obtain a protein with isotopic tags.
Cryo-EM is a simple freezing of protein solution and microscopy. The minus of the method is low resolution (only the general shape of the molecule is visible, but how it is arranged inside is not visible), plus the protein density is close to the density of water / solvent, therefore the signal sinks in a high noise level. In this method, computer technologies for working with pictures and statistics are actively used to pull the signal out of noise.

Millions of protein molecules are selected, classes are divided according to the orientation of the molecule relative to the substrate, class averaging, eigenimages generation, a new round of averaging, and so on until it converges. Then from the information from different classes you can restore the three-dimensional view of the molecule with a low resolution. If there is an internal symmetry of the particles (for example, during cryo-EM analysis of viruses), then each particle can be averaged according to the symmetry operators - then the resolution will be even better, but worse than in the case of X-ray analysis.
X-ray diffraction analysis is the main method for determining protein structures. The main advantage is that it is potentially possible to obtain crystals of even very large complexes from many dozens of proteins (for example, this is how the structure of the ribosome was determined - the Nobel Prize of 2009). The minus of the method - you first need to get a protein crystal, but not every protein wants to crystallize.
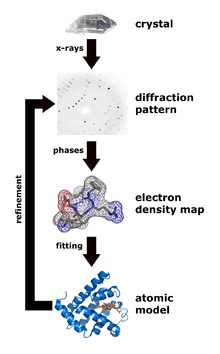
But after the crystal is obtained, the x-ray diffraction can unambiguously determine the positions of all (ordered) atoms in the protein molecule, this method gives the highest resolution and allows you to see the positions of individual atoms in the best cases. It was proved that the structure of the protein in the crystal is uniquely consistent with the structure in solution.
Now the convention is in effect - if you determined the structure of the protein by any of the experimental physical methods, the structure should be placed in open access to the protein data bank (Protein Data Bank - PDB, www.pdb.org ), currently there are more than 90,000 structures (however, many of them are repetitive, for example, complexes of the same protein with different small molecules, such as drugs). In PDB, all structures are in a standard format called, suddenly, pdb. This is a text format in which each atom of the structure corresponds to one line, in which the number of the atom in the structure, the name of the atom (carbon, nitrogen, etc.), the name of the amino acid that includes the atom, the name of the protein chain (A, B, C, etc.) , if it is a crystal of a complex of several proteins), the number of amino acids in the chains and the three-dimensional coordinates of the atom in angstroms relative to the origin, plus the so-called temperature factor and population (these are purely crystallographic parameters).
Then there are special programs that, according to the data from this text file, can graphically display the beautiful three-dimensional structure of the protein molecule, which can be twisted on the monitor screen and, as Guy Dodson said, “touch the molecule with the mouse” (for example, PyMol , CCP4mg , old RasMol ) . That is, it is easy to look at protein structures - you put the program, load the necessary structure from the PDB and enjoy the beauty of nature.
So, we understand the basic idea: protein is a linear polymer that collapses in an aqueous solution under the action of many weak interactions into a stable and unique three-dimensional structure for this protein, and is capable of performing its function in this form. There are several levels of organization of protein structures. Above, we have already become acquainted with the primary structure - a linear sequence of amino acids, which can be written out in a line.
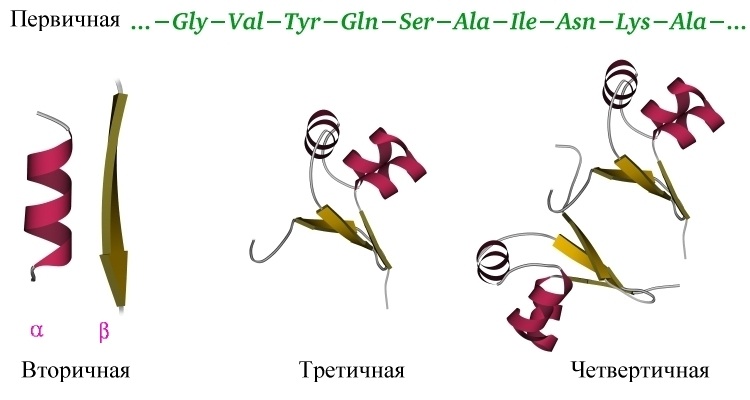
The secondary structure of the protein is determined by the interaction of the atoms of the main protein chain. As mentioned above, the main chain of the protein includes donors and hydrogen bond acceptors, thus, the main chain can acquire some structure. More precisely, several different structures (details nevertheless depend on differing side groups), since the formation of different alternative hydrogen bonds between the main chain groups is possible. Structures are such: alpha-helix, beta sheets (consisting of several beta strands), which are parallel and anti-parallel, beta turn. Plus, part of the chain may not have a pronounced structure, for example, in the region of the rotation of the protein loop. These types of structures have their well-established schematics - an alpha-helix in the form of a spiral or a cylinder, beta-strands in the form of wide arrows. The secondary structure can be reliably predicted by the primary (the standard is JPred ), the alpha-helix is predicted most accurately, with beta strands are lining.
The tertiary structure of the protein is determined by the interaction of the side groups of amino acid residues, this is the three-dimensional structure of the protein. One can imagine that the secondary structure is formed and now these spirals and beta strands want to fit together in a compact three-dimensional structure so that all hydrophobic side groups calmly “stick together” together in the depths of the protein globule, forming a hydrophobic core, and the polar and charged residues stuck out out into the water, forming the surface of the protein and stabilizing the contacts between the elements of the secondary structure. The tertiary structure is schematically depicted in several ways. If you just draw all the atoms, you get porridge (although when we analyze the active center of the protein, we want to look at all the atoms of the active residues).
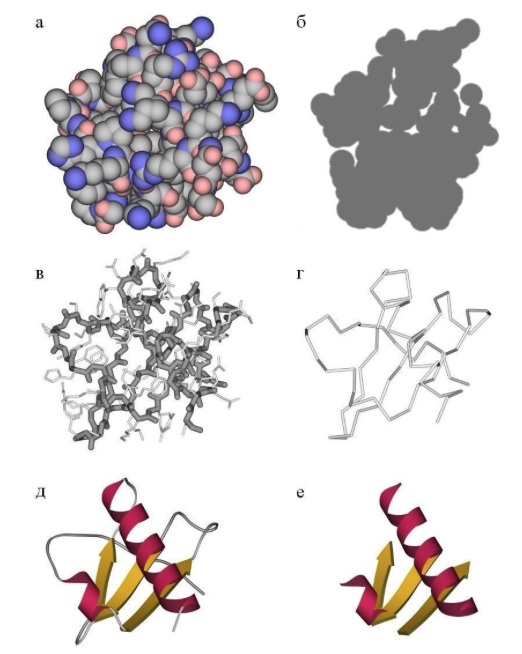
If we want to see how the whole protein works in general, we can display only some atoms of the main chain to see its progress. Alternatively, you can draw a beautiful scheme, where elements of the secondary structure are schematically drawn over the real arrangement of atoms - this is how the protein is seen at a glance. After studying the entire structure in a general, schematic form, you can display the chemical groups of the active center and focus on them. The task of predicting the tertiary structure of a protein is nontrivial and, in general, cannot be solved, although it can be solved in particular cases. More details below.
The quaternary structure of the protein - yes, there is one, though not all proteins. Many proteins work by themselves (monomers, in this case, the monomer means a single folded polypeptide chain, that is, the whole protein), then their quaternary structure is equal to the tertiary one. However, a lot of proteins work only in a complex consisting of several polypeptide chains (subunits or monomers - dimers, trimers, tetramers, multimers), then this assembly of several separate chains is called a quaternary structure. The most commonplace example is hemoglobin consisting of 4 subunits , the most beautiful example in my opinion is the bacterial protein TRAP consisting of 11 identical subunits.
Protein is a complex system of thousands of atoms, so without using computers in the structure of the protein can not figure out. There are many tasks, both solved at an acceptable level and completely unsolved. I will list the most relevant:
At the level of the primary structure - the search for proteins with a similar amino acid sequence, the construction of evolutionary trees on them, and so on - the classic tasks of bioinformatics. The main hub is NCBI - The National Center for Biotechnology Information, www.ncbi.nlm.nih.gov . BLAST is standardly used to search for proteins with a similar sequence: blast.ncbi.nlm.nih.gov/Blast.cgi
Prediction of protein solubility. The point is that if we read the genome of an animal, determine protein sequences using it, deflect these genes into E. coli or the baculovirus expression system, then it turns out that when expressed in these systems, about a third of the proteins will not fold into the correct structure. and, as a result, will be insoluble. It turns out that large proteins actually consist of separate “domains”, each of which represents an autonomous, functional part of a protein (carrying one of its functions) and often “cutting out” a separate domain from a gene, you can get a soluble protein, determine its structure and experiment with it. People try to use machine learning (neural networks, SVM and other classifiers) to predict protein solubility, but it works quite badly (Google will show a lot of things on request to “protein solubility prediction” - there are many servers, but in my experience they all work disgustingly on my squirrels). Ideally, I would like to see a service that would reliably say where those soluble domains are in the protein so that they can be cut out and work with them - there is no such service.
At the level of the secondary structure - the prediction of the same secondary structure by the primary ( JPred )
At the level of the tertiary structure - the search for proteins with similar three-dimensional structures ( DALI , en.wikipedia.org/wiki/Structural_alignment ),
Search for structures in a given sub-structure. For example, I have an arrangement of three amino acids of the active center in space. I want to find structures that contain the same three amino acids in the same relative arrangement, or find structures of proteins, mutation of which will make it possible to arrange the necessary amino acids in the right way. (google "protein substructure search")
Prediction of potential mobility of a three-dimensional structure, possible conformational changes - normal mode analysis, ElNemo .
At the level of the quaternary structure - suppose the structures of the two proteins are known. It is known that they form a complex. Predict the structure of the complex (determine how these two proteins will interact through shape matching, for example). Google "protein-protein docking"
He singled out this computational problem into a separate section, because it is large, fundamental and not solved in the general case.
Experimentally, we know that if you take a protein, fully deploy it and throw it into water, it will roll back to its original state within a time from milliseconds to seconds (this statement is true at least for small globular proteins without any pathologies). This means that all the information needed to determine the three-dimensional structure of a protein is implicitly contained in its primary sequence, so you want to learn how to predict the three-dimensional structure of a protein by the amino acid sequence in silico ! However, this problem is generally not solved so far. What is the matter? The fact is that in the primary sequence there is no explicit information necessary for the construction of the structure. Firstly, there is no information about the conformation of the main chain - and it has significant mobility, although it is somewhat limited for steric reasons. Plus, each side chain of each amino acid may be in different conformations, for long side groups such as arginine, it may be more than a dozen conformations.
What to do? There is a fairly well-known habravchanam the most common approach, called "molecular dynamics" and suitable for all molecules and systems. , , , , , . ? , , . – , , – . , . , , ( http://en.wikipedia.org/wiki/Anton_(computer) )! However, it is too early to celebrate victory. They took a very small (its size is 5-10 times less than the average protein) and one of the fastest-folding proteins, the classic model protein, on which folding was studied. For large proteins, the calculation time increases nonlinearly and will take years, that is, there is still something to work on.
Another approach is implemented in Rosetta . (3-9 ) , PDB, - , . - , , : « , - ?».
– , . FoldIt , , - ( – !). , CASP . , , PDB, PDB, CASP . , , , . , FoldIt , , - CASPamong professionals modeling protein structures and predicted protein structure more accurately. However, even these successes do not suggest that the problem of predicting the structure of the protein is solved - very often the model is very far from the real structure.
All this concerned ab initio protein modeling. , . , PDB . . , 30% ( , ). ( ) , « », «» , , - . , – . – Modeller , SwissModel .
You can solve other problems, for example, try to model what will happen if you bring this or that mutation into the protein. For example, if you replace the hydrophilic amino acid on the surface of a protein with another hydrophilic one, then most likely the structure of the protein will not change at all. If you replace the amino acid from the hydrophobic nucleus with another hydrophobic one, but of a different size, then most likely the protein placement will remain the same, but will slightly “eat up” by an angstrom. If we replace the amino acid from the hydrophobic nucleus with the charged one, then most likely the protein simply “explodes” and cannot coagulate.
, . , -, , – « » . « ?», « ?», « ?».
: , . , , . 2-3 , . – 3 . , . , , «/» , , . , . , « , ».
, , , .
, , .
:
1) « » . , : phys.protres.ru/lectures/protein_physics/index.html ( )
2) , « », II « ». Djvu
3) , , PubMed ( www.pubmed.org ) - he should be asked to read about "protein engineering" and the like.
1. Why are proteins important?
As Friedrich Engels said, “Life is a way of existence of protein bodies”. In the 19th century, they still did not know about the role of DNA in the inheritance of genetic information, but Uncle Frederick’s statement is still largely true - proteins do most of the work in our cells. This includes maintaining the structure (cell shape), chemical catalysis, and motor function (muscle contraction, for example), and transport (say, hemoglobin protein transports oxygen from the lungs to the tissue and carbon dioxide in the opposite direction) and complex regulatory functions to maintain consistency internal environment (for example, protein hormones and all intracellular regulatory systems) and many others. In a word, if something happens in our body, proteins are necessarily involved (though not only them).
2. What is protein?
From a chemical point of view, a protein is a linear (unbranched) polymer consisting of monotonously repeating identical blocks of the “main chain”, to which various “side groups” are attached. Since the main chain blocks are asymmetric, the entire polypeptide chain of the protein has a direction, the N- and C-termini of the polypeptide chain are distinguished.
The chain length is from 70 to more than 1000 monomers (amino acid residues), the average length for higher organisms is about 500-600 amino acid residues, for bacteria this value will be less, most likely 300-400 residues. In total, there are 20 standard amino acids in nature, the same for both bacteria and humans, that is, 20 different side groups can stick out from the main chain.
(A correction is possible here - some chemical groups can be modified after protein synthesis, for example, phosphorylated. However, this is not considered as another amino acid, but is considered as a product of the original modification. Also, higher non-canonical amino acids are possible in higher organisms, but this is a rare event. That is, strictly speaking, 22 different amino acids, 20 of them are basic and 2 rare, plus some side groups can be rarely chemically modified).
From generation to generation, genetic information is transmitted in the form of DNA, there are so-called “protein-coding regions” in it. In these places, DNA unambiguously (for botans - up to alternative splicing and RNA editing) encoded information about the linear amino acid sequence for the synthesis of this protein, plus there are corresponding machines in the cell that can synthesize a protein according to information originally encoded in DNA.
')
Since protein is a linear polymer assembled from 20 standard monomers, its so-called “primary structure” can be easily represented as a string, for example:
> small ubiquitin-related modifier 3 precursor [Homo sapiens] MSEEKPKEGVKTENDHINLKVAGQDGSVVQFKIKRHTPLSKLMKAYCERQG LSMRQIRFRFDGQPINETDTPAQLEMEDEDTIDVFQQQTGGVPESSLAGHSF
This is the amino acid sequence of a small human protein in the FASTA format, the first line starting with “>” describes its name, followed by the amino acid sequence in accordance with the standard coding (for example, M is methionium, S is serine, etc., a total of 20 letters of standard single-letter code), on the left is the N-terminus of the protein, on the right is its C-terminus. For different proteins, the length of the string will be obviously different, since proteins have different lengths. The sequences of all known proteins can be found in the public domain here: www.ncbi.nlm.nih.gov
3. Protein Structure
Well, the primary structure is sorted out, but does the protein work in an expanded linear form? Of course not. Here it should be noted that from a structural point of view there are different classes of proteins: globular, membrane and fibrillar. Membrane proteins, as the name implies, live only in cell membranes, to stabilize their structure, you need a special membrane environment, we will not consider them in this review. Fibrillar proteins have a simple regular structure, look like elongated fibers, they are insoluble in water and perform structural functions (for example, hair consists of keratin, fibrillar proteins include protein from natural silk). Recently, they began to distinguish a class of disordered proteins — proteins that do not have a constant three-dimensional structure, or that acquire it only for a short time when interacting with other proteins. The most interesting from a practical point of view, the class of proteins, which we will consider - globular water-soluble proteins, most proteins belong to this class.
A linear polypeptide chain in water is capable of spontaneously folding into a complex three-dimensional structure (globule) and only in such a folded form can proteins perform chemical catalysis and other interesting work. Therefore, it is fundamentally important for us to know exactly the three-dimensional folding of the protein, since it is only at this level that it becomes clear how the protein works.
Question : how many three-dimensional structures correspond to a specific protein?
Answer : One, up to a small mobility of small “disordered” loops. Only one exception is known, when two sufficiently different structures correspond to one sequence, these are prions .
Question : Why does the protein have only one three-dimensional structure?
Answer : for chemical catalysis, we need to arrange the corresponding chemical groups in a strictly defined way in space. This requires a rigid structure. That is, the whole protein must be rigid in order to maintain the chemical groups of amino acids of the active center in the right places (in reality, many proteins consist of two or more rigid parts that can move relative to each other, this is necessary for regulating the activity of the protein ( allosteric regulation ) so that a certain signal could turn on and off the chemical activity of the protein-enzyme). To make the structure rigid and stable, nature made sure that the structure of each protein corresponded to the energy minimum of this system of atoms and this minimum was so deep that the protein did not “jump out” from it. All other parasitic structures have more energy and the protein still falls into the energy minimum corresponding to the native structure.
Question : what keeps the three-dimensional structure of the protein?
Answer : In short, mainly on a large number of non-covalent interactions. In principle, chemical groups of a protein can form: (1) a hydrogen bond, these groups exist in the main chain and in some side groups, (2) an ionic bond is an electrostatic interaction between oppositely charged side groups, (3) Van der Waalsovo interaction and (4) the hydrophobic effect, which keeps the overall structure of the protein. The point is that there are always hydrophobic aromatic residues in a protein, it is energetically unprofitable for them to contact with polar water molecules, and it is advantageous to “stick together” with each other. Thus, when the protein is folded, the hydrophobic groups are pushed out of the water environment, “sticking together” with each other and forming a “hydrophobic core”, while the polar and charged groups, on the contrary, tend to the water environment, forming the surface of the protein globule. Also (5), the side groups of two cysteine residues can form a disulfide bridge between them — a full-fledged covalent bond that rigidly fixes the protein.
Accordingly, all amino acids are divided into hydrophobic, polar (hydrophilic), positively and negatively charged. Plus cysteines capable of forming a covalent bond between each other. Glycine has special properties - it does not have a side group that severely restricts the conformational mobility of other residues, so it can “bend” very much and is located in places where the protein chain must be unfolded. In Proline, on the contrary, the side group forms a ring covalently linked to the main chain, rigidly fixing its conformation. Prolines are found where it is necessary to make the protein chain rigid and stiff. Many diseases are associated with the mutation of proline to glycine, due to which the structure of the protein slightly “floats”.
Question : how do we even know about the three-dimensional structures of the protein?
Answer : from the experiment, this is absolutely reliable data.
Now there are 3 methods for experimentally determining the structure of a protein: nuclear magnetic resonance (NMR), cryo-EM (electron microscopy), and x-ray diffraction analysis of protein crystals.
NMR allows you to determine the structure of the protein in solution, but it works only for very small proteins (for large ones it is impossible to make deconvolution).
This method was important for general evidence that a protein has only one three-dimensional structure and that the structure of a protein in a crystal is identical to the structure in solution. This is a very expensive method, since it is required to obtain a protein with isotopic tags.
Cryo-EM is a simple freezing of protein solution and microscopy. The minus of the method is low resolution (only the general shape of the molecule is visible, but how it is arranged inside is not visible), plus the protein density is close to the density of water / solvent, therefore the signal sinks in a high noise level. In this method, computer technologies for working with pictures and statistics are actively used to pull the signal out of noise.
Millions of protein molecules are selected, classes are divided according to the orientation of the molecule relative to the substrate, class averaging, eigenimages generation, a new round of averaging, and so on until it converges. Then from the information from different classes you can restore the three-dimensional view of the molecule with a low resolution. If there is an internal symmetry of the particles (for example, during cryo-EM analysis of viruses), then each particle can be averaged according to the symmetry operators - then the resolution will be even better, but worse than in the case of X-ray analysis.
X-ray diffraction analysis is the main method for determining protein structures. The main advantage is that it is potentially possible to obtain crystals of even very large complexes from many dozens of proteins (for example, this is how the structure of the ribosome was determined - the Nobel Prize of 2009). The minus of the method - you first need to get a protein crystal, but not every protein wants to crystallize.
But after the crystal is obtained, the x-ray diffraction can unambiguously determine the positions of all (ordered) atoms in the protein molecule, this method gives the highest resolution and allows you to see the positions of individual atoms in the best cases. It was proved that the structure of the protein in the crystal is uniquely consistent with the structure in solution.
Now the convention is in effect - if you determined the structure of the protein by any of the experimental physical methods, the structure should be placed in open access to the protein data bank (Protein Data Bank - PDB, www.pdb.org ), currently there are more than 90,000 structures (however, many of them are repetitive, for example, complexes of the same protein with different small molecules, such as drugs). In PDB, all structures are in a standard format called, suddenly, pdb. This is a text format in which each atom of the structure corresponds to one line, in which the number of the atom in the structure, the name of the atom (carbon, nitrogen, etc.), the name of the amino acid that includes the atom, the name of the protein chain (A, B, C, etc.) , if it is a crystal of a complex of several proteins), the number of amino acids in the chains and the three-dimensional coordinates of the atom in angstroms relative to the origin, plus the so-called temperature factor and population (these are purely crystallographic parameters).
ATOM 1 N HIS A 17-12.690 8.753 5.446 1.00 29.32 N ATOM 2 CA HIS A 17 -11.570 8.953 6.350 1.00 21.61 C ATOM 3 C HIS A 17-10.274 8.970 5.544 1.00 22.01 C ATOM 4 O HIS A 17-10.193 8.315 4.491 1.00 29.95 O ATOM 5 CB HIS A 17-11.462 7.820 7.380 1.00 23.64 C ATOM 6 CG HIS A 17-12.551 7.811 8.421 1.00 21.18 C ATOM 7 ND1 HIS A 17-13.731 7.137 8.194 1.00 28.94 N ATOM 8 CD2 HIS A 17-12.634 8.384 9.644 1.00 21.69 C ATOM 9 CE1 HIS A 17-14.492 7.301 9.267 1.00 27.01 C ATOM 10 NE2 HIS A 17-13.869 8.058 10.168 1.00 22.66 N ATOM 11 N ILE A 18 -9.269 9.660 6.089 1.00 19.45 N ATOM 12 CA ILE A 18-7.710 9.377 5.605 1.00 18.67 C ATOM 13 C ILE A 18-7.122 8.759 6.749 1.00 16.24 C ATOM 14 O ILE A 18-7.425 8.919 7.929 1.00 18.80 O ATOM 15 CB ILE A 18-7.228 10.640 5.088 1.00 20.22 C ATOM 16 CG1 ILE A 18-7.062 11.686 6.183 1.00 18.52 C ATOM 17 CG2 ILE A 18-7.781 11.176 3.889 1.00 24.61 C ATOM 18 CD1 ILE A 18-6.161 12.824 5.749 1.00 28.21 C ATOM 19 N ASN A 19-6.121 8.023 6.349 1.00 15.46 N ATOM 20 CA ASN A 19 -5.239 7.306 7.243 1.00 14.34 C ATOM 21 C ASN A 19-4.012 8.178 7.507 1.00 14.83 C ATOM 22 O ASN A 19-3.431 8.715 6.575 1.00 18.03 O ATOM 23 CB ASN A 19-4.825 6.003 6.573 1.00 17.71 C ATOM 24 CG ASN A 19-6.062 5.099 6.413 1.00 21.26 C ATOM 25 OD1 ASN A 19,66.606 4.651 7.400 1.00 26.18 O ATOM 26 ND2 ASN A 19-6.320 4.899 5.151 1.00 31.73 N
Then there are special programs that, according to the data from this text file, can graphically display the beautiful three-dimensional structure of the protein molecule, which can be twisted on the monitor screen and, as Guy Dodson said, “touch the molecule with the mouse” (for example, PyMol , CCP4mg , old RasMol ) . That is, it is easy to look at protein structures - you put the program, load the necessary structure from the PDB and enjoy the beauty of nature.
4. Analyzing the structure
So, we understand the basic idea: protein is a linear polymer that collapses in an aqueous solution under the action of many weak interactions into a stable and unique three-dimensional structure for this protein, and is capable of performing its function in this form. There are several levels of organization of protein structures. Above, we have already become acquainted with the primary structure - a linear sequence of amino acids, which can be written out in a line.
The secondary structure of the protein is determined by the interaction of the atoms of the main protein chain. As mentioned above, the main chain of the protein includes donors and hydrogen bond acceptors, thus, the main chain can acquire some structure. More precisely, several different structures (details nevertheless depend on differing side groups), since the formation of different alternative hydrogen bonds between the main chain groups is possible. Structures are such: alpha-helix, beta sheets (consisting of several beta strands), which are parallel and anti-parallel, beta turn. Plus, part of the chain may not have a pronounced structure, for example, in the region of the rotation of the protein loop. These types of structures have their well-established schematics - an alpha-helix in the form of a spiral or a cylinder, beta-strands in the form of wide arrows. The secondary structure can be reliably predicted by the primary (the standard is JPred ), the alpha-helix is predicted most accurately, with beta strands are lining.
The tertiary structure of the protein is determined by the interaction of the side groups of amino acid residues, this is the three-dimensional structure of the protein. One can imagine that the secondary structure is formed and now these spirals and beta strands want to fit together in a compact three-dimensional structure so that all hydrophobic side groups calmly “stick together” together in the depths of the protein globule, forming a hydrophobic core, and the polar and charged residues stuck out out into the water, forming the surface of the protein and stabilizing the contacts between the elements of the secondary structure. The tertiary structure is schematically depicted in several ways. If you just draw all the atoms, you get porridge (although when we analyze the active center of the protein, we want to look at all the atoms of the active residues).
If we want to see how the whole protein works in general, we can display only some atoms of the main chain to see its progress. Alternatively, you can draw a beautiful scheme, where elements of the secondary structure are schematically drawn over the real arrangement of atoms - this is how the protein is seen at a glance. After studying the entire structure in a general, schematic form, you can display the chemical groups of the active center and focus on them. The task of predicting the tertiary structure of a protein is nontrivial and, in general, cannot be solved, although it can be solved in particular cases. More details below.
The quaternary structure of the protein - yes, there is one, though not all proteins. Many proteins work by themselves (monomers, in this case, the monomer means a single folded polypeptide chain, that is, the whole protein), then their quaternary structure is equal to the tertiary one. However, a lot of proteins work only in a complex consisting of several polypeptide chains (subunits or monomers - dimers, trimers, tetramers, multimers), then this assembly of several separate chains is called a quaternary structure. The most commonplace example is hemoglobin consisting of 4 subunits , the most beautiful example in my opinion is the bacterial protein TRAP consisting of 11 identical subunits.
5. Computational problems
Protein is a complex system of thousands of atoms, so without using computers in the structure of the protein can not figure out. There are many tasks, both solved at an acceptable level and completely unsolved. I will list the most relevant:
At the level of the primary structure - the search for proteins with a similar amino acid sequence, the construction of evolutionary trees on them, and so on - the classic tasks of bioinformatics. The main hub is NCBI - The National Center for Biotechnology Information, www.ncbi.nlm.nih.gov . BLAST is standardly used to search for proteins with a similar sequence: blast.ncbi.nlm.nih.gov/Blast.cgi
Prediction of protein solubility. The point is that if we read the genome of an animal, determine protein sequences using it, deflect these genes into E. coli or the baculovirus expression system, then it turns out that when expressed in these systems, about a third of the proteins will not fold into the correct structure. and, as a result, will be insoluble. It turns out that large proteins actually consist of separate “domains”, each of which represents an autonomous, functional part of a protein (carrying one of its functions) and often “cutting out” a separate domain from a gene, you can get a soluble protein, determine its structure and experiment with it. People try to use machine learning (neural networks, SVM and other classifiers) to predict protein solubility, but it works quite badly (Google will show a lot of things on request to “protein solubility prediction” - there are many servers, but in my experience they all work disgustingly on my squirrels). Ideally, I would like to see a service that would reliably say where those soluble domains are in the protein so that they can be cut out and work with them - there is no such service.
At the level of the secondary structure - the prediction of the same secondary structure by the primary ( JPred )
At the level of the tertiary structure - the search for proteins with similar three-dimensional structures ( DALI , en.wikipedia.org/wiki/Structural_alignment ),
Search for structures in a given sub-structure. For example, I have an arrangement of three amino acids of the active center in space. I want to find structures that contain the same three amino acids in the same relative arrangement, or find structures of proteins, mutation of which will make it possible to arrange the necessary amino acids in the right way. (google "protein substructure search")
Prediction of potential mobility of a three-dimensional structure, possible conformational changes - normal mode analysis, ElNemo .
At the level of the quaternary structure - suppose the structures of the two proteins are known. It is known that they form a complex. Predict the structure of the complex (determine how these two proteins will interact through shape matching, for example). Google "protein-protein docking"
6. Prediction of protein structure
He singled out this computational problem into a separate section, because it is large, fundamental and not solved in the general case.
Experimentally, we know that if you take a protein, fully deploy it and throw it into water, it will roll back to its original state within a time from milliseconds to seconds (this statement is true at least for small globular proteins without any pathologies). This means that all the information needed to determine the three-dimensional structure of a protein is implicitly contained in its primary sequence, so you want to learn how to predict the three-dimensional structure of a protein by the amino acid sequence in silico ! However, this problem is generally not solved so far. What is the matter? The fact is that in the primary sequence there is no explicit information necessary for the construction of the structure. Firstly, there is no information about the conformation of the main chain - and it has significant mobility, although it is somewhat limited for steric reasons. Plus, each side chain of each amino acid may be in different conformations, for long side groups such as arginine, it may be more than a dozen conformations.
What to do? There is a fairly well-known habravchanam the most common approach, called "molecular dynamics" and suitable for all molecules and systems. , , , , , . ? , , . – , , – . , . , , ( http://en.wikipedia.org/wiki/Anton_(computer) )! However, it is too early to celebrate victory. They took a very small (its size is 5-10 times less than the average protein) and one of the fastest-folding proteins, the classic model protein, on which folding was studied. For large proteins, the calculation time increases nonlinearly and will take years, that is, there is still something to work on.
Another approach is implemented in Rosetta . (3-9 ) , PDB, - , . - , , : « , - ?».
– , . FoldIt , , - ( – !). , CASP . , , PDB, PDB, CASP . , , , . , FoldIt , , - CASPamong professionals modeling protein structures and predicted protein structure more accurately. However, even these successes do not suggest that the problem of predicting the structure of the protein is solved - very often the model is very far from the real structure.
All this concerned ab initio protein modeling. , . , PDB . . , 30% ( , ). ( ) , « », «» , , - . , – . – Modeller , SwissModel .
You can solve other problems, for example, try to model what will happen if you bring this or that mutation into the protein. For example, if you replace the hydrophilic amino acid on the surface of a protein with another hydrophilic one, then most likely the structure of the protein will not change at all. If you replace the amino acid from the hydrophobic nucleus with another hydrophobic one, but of a different size, then most likely the protein placement will remain the same, but will slightly “eat up” by an angstrom. If we replace the amino acid from the hydrophobic nucleus with the charged one, then most likely the protein simply “explodes” and cannot coagulate.
, . , -, , – « » . « ?», « ?», « ?».
: , . , , . 2-3 , . – 3 . , . , , «/» , , . , . , « , ».
, , , .
, , .
:
1) « » . , : phys.protres.ru/lectures/protein_physics/index.html ( )
2) , « », II « ». Djvu
3) , , PubMed ( www.pubmed.org ) - he should be asked to read about "protein engineering" and the like.
Source: https://habr.com/ru/post/181850/
All Articles