Darwinian Evolution of Bacteria - The Complete Picture
I will start with a provocative statement - "biologists do not publish the details of their research." It would seem that there are so many articles, so many studies ... but where is the description and detail of the information that is obtained? It is not in principle. And articles without such information are empty and controversial. Everyone praises his method, but how many people have bothered to verify the data of others, and most importantly, could he do it?
We can only welcome the appearance of such bio-information bases as the NCBI genomes and PDB , in which researchers place data on the sequenced genomes and structures of RNA and proteins. And most importantly, some scientists, before publishing an article, first put the data into bioinformatics databases.
You tell me there are many other bases - but I’ll tell you they are less serious, and as a rule these are two posts with some adaptation. But the main thing is that all other bio-informational information, it may be said secondary, does not fit into the bases. And in the articles, however, there are various speculations.
')
Of course, it looks like this only for amateurs like me. Real professionals have everything in a pharmacy. Therefore, you can not bother to answer these pretentious statements. We'll just talk about what bioinformatics looks like in its private areas through the eyes of an amateur. But maybe this story will lead you to something.
We will discuss below the construction of an evolutionary tree according to Darwin, let's look at how much this is true and I will eventually give a complete tree (within the available information) of the evolution of bacteria based on the most conservative tRNA genes. And I will give an explanation about the method of constructing such a tree.
Experts in bioinformatics recommend reading from section number 5, skipping all my pathos.
Being an amateur, I always wondered about one thing - how can organisms be classified and systematized when there is no information about DNA, when strains of organisms are not yet sequenced? And please, the Burgey Handbook only in the last edition began to take into account information about genes. And before that, it took into account only the structural and functional characteristics of bacteria.
I'm not talking about those conservative biologists who say in all seriousness that taxonomy should be built not only on the basis of a genomic comparison, but also on the basis of morphological and physiological data. And this is the gene century, we must return to the time of C. Linnaeus?
But in the absence of a more authoritative publication than the Burgey Handbook, bio-information bases on taxonomy, such as in the NCBI , are more complete and sometimes have links to sequenced strains — the principle of building such a tree is just a repost of the Burgey reference book.
Say no ... ok, you can easily find the difference. But you will never understand why a tree is exactly what it is. It is certainly attributed to this or that species, who gave such a name to the taxon, and if the article is lucky, and if it’s very lucky, the article will briefly describe why this taxon was placed one way or another in systematics.
Further, if we take individual articles on the construction of phylogenetic trees - in them, at best, a very small number of species are considered, and the trees are built using completely transparent methods and not large enough.
There are many professionals who try to make it so that the problem of an amateur is his lack of education and under-awareness.
This is partly true, but only partly. Amateurs are not doing their job, because having their profession - they are also interested in things from others and think in what other area they can apply their knowledge. And when they see something like this as I described above for taxonomy, they are confused.
They take the most naive method, since they need a result, not a reason to write an article, and build an evolution tree. Then the professionals begin to resent how so - they are doing it professionally, but there are no results ... the grants are not all used. Although it is possible to take one person to build it all without any particular difficulties and without scoring his head with methods in which complexity is introduced for the sake of complexity. And so it turns out the result of the amateur.
It can be discussed, but it can be discussed seriously only when professionals have at least something comparable and equally transparent. And now we come to this.
Who has read my previous articles knows that I have already written on this topic since the article Interesting results about the evolutionary systematics of prokaryotes or “multiple species” , and not so long ago gave more complete results in the article Systematics of prokaryotes - distant relatives . Here I would like to tell you how my worldview has changed as this study progressed.
Initially, the article showed that on the basis of one type of tRNA that transferred alanine, one can find a stable connection between different species, genera, etc. I interpreted this connection as sexual inheritance, since it was possible to find organisms in which tRNA_Ala existed from two other species. There were some exceptions, but they were relatively few. “What could be simpler,” the amateur exclaimed, “it's the same genes from mom and dad, and biologists fool us with asexual reproduction.”
My critics hardly noticed this idea at that time (apparently writing off the horizontal transfer - although the mum-dad communication was very constant), but noted that it was not serious to draw conclusions based on one gene.
I readily agreed, but I thought to myself - how many genes do you analyze yourself? Correctly, as a rule, one 16S, only it will be longer, but it will be rugged with mutations. But what can we compare with others ... go ahead.
Then I took all the sequenced genomes and all the tRNAs available in the NCBI and clarified the information (see. Systematics of prokaryotes - distant relatives ).
Critics did not become less, but she became more emotional. Yeah, I thought it becomes harder to object, and the arguments of opponents are far from being considered and indirect.
But I saw that the whole picture became very confusing, there was a feeling that the genera interact somewhere weaker, somewhere stronger - but almost like everyone with each. One or another type of gene they had in common.
Imagine that this could really be evolution. as if all the genes were thrown into one pot, and then they would scoop a random set from this pot and create a look — it was somehow difficult. But the results talked about it inexorably.
Different genera, although slightly distinguished in groups, looked as though tRNA were transferred horizontally in a random way.
In fact, there is no 100% reason not to believe in “multiple origin”. This is exactly the same speculation as Darwinian evolution. These speculations should be explicitly called methods of interpreting experimental data. “Multicultural origin” is a display of a graph on which there are exact links on genes between genera.
But in this graph there is no direction of evolution, this graph makes no assumptions about the past. It simply shows the facts of kinship of modern organisms. At the same time the relationship of these organisms can be distant and on the basis of this graph it is not possible to tell when the divergence of species occurred.
Darwinian evolution is another way of interpretation, which makes it possible to imagine the most detailed course of evolution.
But here the amateur faced again with bewilderment from classical ideas, or rather just from the lack of results. I was told by an opponent that such a concept as “ancient” is bad for biology, since based on the available methods, it is impossible to estimate the relative time of occurrence of the species. But after all, after clarifying a number of points, we all agreed on the following:
This is what I called the interpretation of Darwinian evolution. But let me specially note that although all Darwinists (that is, classical taxonomists and phylogenetics) should deal with this, they build trees using measures that are more similar to interpreting "many-species origin", and of course it is difficult for them to talk about "antiquity of the species" »By definition, such an interpretation - as mentioned above, there is no direction of evolution and there can be no.
But the opponent turned out to be wrong in his assessment “I think that there will not be much difference between the different types” - it is and significant, and this will be demonstrated further - just look at the resulting tree of evolution.
From here can read those who are squeamish about the pathetic text of the amateur that is above.
To understand it is necessary to read the article Systematics of prokaryotes - distant relatives , it describes the basics, which are the input data. Explaining further, I assume that you understand what this graph means, for example, and how it was constructed:
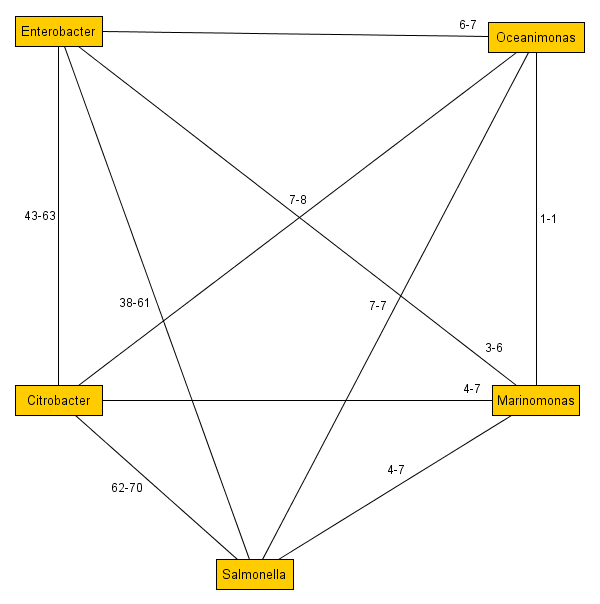
Now we need to figure out how to transform it into a tree with directional evolution, for example:

In this tree we restore the ancestors of the modern genera of bacteria. Modern sorts of bacteria have names and are represented as leaves of a tree, while their ancestors are indicated by a set of numbers.
Each digit is the identifier of the tRNA group that an ancestor must possess in order to pass on to its descendants in the next generation. If he did not possess such a group of tRNA, then we definitely could not get the current state of the relationship (coincidence of identical tRNA), which is available in the column of "multiple origin" above.
Thus, the algorithm for constructing such a tree consists of two parts:
1. Distribution of tRNA into groups, so that the entire analyzed set can be appealed only in groups without switching to single tRNA — this is necessary for two purposes (1) it is an order of magnitude more convenient to deal with groups than with a large set of tRNA. Duplicate information is eliminated, and the group is the minimum unit of divergence. (2) Groups can be sorted by the number of tRNAs entering there. The probability of divergence (separation) of a larger group in different genera is higher with a smaller number of ancestral divergences (branch length).
2. Actually building ancestral tree.
Further, I will describe only the general principle of the implementation of these two parts.
Division into groups:
1. At the entrance there is information of the form:
1 10 000913,003420,00686818,011215,013800,016316,017374,
2 9 000434,000487,005891,005892,011142,01111163,
2 10 000913,003420,006868,007509,011215,013800,016316,017374,
2 8 000487,003420,005891,006678,011163,013218,007509,
it describes a graph of “multi-species origin”, namely a set of links, where “1” is the identification of one kind, “10” is the identification of the second kind, “000913,003420,00686818.011215.013800,016316.017374,” are those tRNAs that identical in both the first and second kind.
2. The first group is created, as a set of all generally different tRNAs.
3. Distribution into groups takes place, if tRNA on relationships between genera belongs to a group, this set is replaced with a group identification, but if the occurrence is partial, then what tRNA is labeled is missing, or, conversely, which tRNA, are only from this group.
4. The division of the group into two. The above made distribution into groups is analyzed, the first partial entry is taken - a new group is created, and the missing part remains with the previous group.
5. Repeating paragraph 3. So gradually, there will be a division into groups without partial occurrences.
6. Groups are sorted by value 1 - a group is, say, a set of 20 tRNAs, and after 300 groups there is an entry of 1-2 tRNA
Ancestral tree construction:
1. Based on the division of the genus links into groups of tRNA, it is possible to recover which groups of tRNA are in each genus. So if between births there is such a connection
1 10 307 | 864 | 867 | 897 | 909 | 911 |
6 10 307 | 862 | 864 | 867 | 897 | 909 | 911 |
This means that groups 307 | 864 | 867 | 897 | 909 | 911 | There is also a 1st kind and a 10th. But the 862 group, for example, has only the 10th and 6th, but not the 1st.
2. All genera do leaves of the tree
3. We take the 1st group (remember that it is the largest, which means it is less fragmented and is younger). We find the smallest common ancestor for all genera that possess this group of tRNA. If there is no such ancestor, create it. If there is, but the smallest common ancestor is not marked with the corresponding identifier of the tRNA group - we label.
4. Repeat p.3. for all groups
Well, the actual result of the tree of the evolution of bacteria can be seen in the picture :
We look in high resolution
PS I understand that I did not provide the actual results of the division into groups, that this tRNA cannot be concretely understood, and the method is described only from a bird's eye view. Really interested, I can provide all the information, but I expect from them that they will try to double-check me at least in something and do not hesitate to make it publicly.
We can only welcome the appearance of such bio-information bases as the NCBI genomes and PDB , in which researchers place data on the sequenced genomes and structures of RNA and proteins. And most importantly, some scientists, before publishing an article, first put the data into bioinformatics databases.
You tell me there are many other bases - but I’ll tell you they are less serious, and as a rule these are two posts with some adaptation. But the main thing is that all other bio-informational information, it may be said secondary, does not fit into the bases. And in the articles, however, there are various speculations.
')
Of course, it looks like this only for amateurs like me. Real professionals have everything in a pharmacy. Therefore, you can not bother to answer these pretentious statements. We'll just talk about what bioinformatics looks like in its private areas through the eyes of an amateur. But maybe this story will lead you to something.
We will discuss below the construction of an evolutionary tree according to Darwin, let's look at how much this is true and I will eventually give a complete tree (within the available information) of the evolution of bacteria based on the most conservative tRNA genes. And I will give an explanation about the method of constructing such a tree.
Experts in bioinformatics recommend reading from section number 5, skipping all my pathos.
№1. Taxonomy
Being an amateur, I always wondered about one thing - how can organisms be classified and systematized when there is no information about DNA, when strains of organisms are not yet sequenced? And please, the Burgey Handbook only in the last edition began to take into account information about genes. And before that, it took into account only the structural and functional characteristics of bacteria.
I'm not talking about those conservative biologists who say in all seriousness that taxonomy should be built not only on the basis of a genomic comparison, but also on the basis of morphological and physiological data. And this is the gene century, we must return to the time of C. Linnaeus?
But in the absence of a more authoritative publication than the Burgey Handbook, bio-information bases on taxonomy, such as in the NCBI , are more complete and sometimes have links to sequenced strains — the principle of building such a tree is just a repost of the Burgey reference book.
Say no ... ok, you can easily find the difference. But you will never understand why a tree is exactly what it is. It is certainly attributed to this or that species, who gave such a name to the taxon, and if the article is lucky, and if it’s very lucky, the article will briefly describe why this taxon was placed one way or another in systematics.
Further, if we take individual articles on the construction of phylogenetic trees - in them, at best, a very small number of species are considered, and the trees are built using completely transparent methods and not large enough.
№2. The problem of amateur
There are many professionals who try to make it so that the problem of an amateur is his lack of education and under-awareness.
This is partly true, but only partly. Amateurs are not doing their job, because having their profession - they are also interested in things from others and think in what other area they can apply their knowledge. And when they see something like this as I described above for taxonomy, they are confused.
They take the most naive method, since they need a result, not a reason to write an article, and build an evolution tree. Then the professionals begin to resent how so - they are doing it professionally, but there are no results ... the grants are not all used. Although it is possible to take one person to build it all without any particular difficulties and without scoring his head with methods in which complexity is introduced for the sake of complexity. And so it turns out the result of the amateur.
It can be discussed, but it can be discussed seriously only when professionals have at least something comparable and equally transparent. And now we come to this.
Number 3. Multicultural origin and other nonsense
Who has read my previous articles knows that I have already written on this topic since the article Interesting results about the evolutionary systematics of prokaryotes or “multiple species” , and not so long ago gave more complete results in the article Systematics of prokaryotes - distant relatives . Here I would like to tell you how my worldview has changed as this study progressed.
Initially, the article showed that on the basis of one type of tRNA that transferred alanine, one can find a stable connection between different species, genera, etc. I interpreted this connection as sexual inheritance, since it was possible to find organisms in which tRNA_Ala existed from two other species. There were some exceptions, but they were relatively few. “What could be simpler,” the amateur exclaimed, “it's the same genes from mom and dad, and biologists fool us with asexual reproduction.”
My critics hardly noticed this idea at that time (apparently writing off the horizontal transfer - although the mum-dad communication was very constant), but noted that it was not serious to draw conclusions based on one gene.
I readily agreed, but I thought to myself - how many genes do you analyze yourself? Correctly, as a rule, one 16S, only it will be longer, but it will be rugged with mutations. But what can we compare with others ... go ahead.
Then I took all the sequenced genomes and all the tRNAs available in the NCBI and clarified the information (see. Systematics of prokaryotes - distant relatives ).
Critics did not become less, but she became more emotional. Yeah, I thought it becomes harder to object, and the arguments of opponents are far from being considered and indirect.
But I saw that the whole picture became very confusing, there was a feeling that the genera interact somewhere weaker, somewhere stronger - but almost like everyone with each. One or another type of gene they had in common.
Imagine that this could really be evolution. as if all the genes were thrown into one pot, and then they would scoop a random set from this pot and create a look — it was somehow difficult. But the results talked about it inexorably.
Different genera, although slightly distinguished in groups, looked as though tRNA were transferred horizontally in a random way.
№4. Darwinian evolution - as a way of thinking
In fact, there is no 100% reason not to believe in “multiple origin”. This is exactly the same speculation as Darwinian evolution. These speculations should be explicitly called methods of interpreting experimental data. “Multicultural origin” is a display of a graph on which there are exact links on genes between genera.
But in this graph there is no direction of evolution, this graph makes no assumptions about the past. It simply shows the facts of kinship of modern organisms. At the same time the relationship of these organisms can be distant and on the basis of this graph it is not possible to tell when the divergence of species occurred.
Darwinian evolution is another way of interpretation, which makes it possible to imagine the most detailed course of evolution.
But here the amateur faced again with bewilderment from classical ideas, or rather just from the lack of results. I was told by an opponent that such a concept as “ancient” is bad for biology, since based on the available methods, it is impossible to estimate the relative time of occurrence of the species. But after all, after clarifying a number of points, we all agreed on the following:
I: We can speak of the degree of conservatism of the species as a combination of the presence of conservative molecules closer to luca. That seems to be the difference with us.
Opponent: Yes, I agree with that. that is, if we can restore the “ancestral state” by a large number of genes (which in itself is a rather difficult task), then for each specific species we will be able to determine how close it is to this ancestral state. I think that there will not be much difference between different species, but definitely there is, for whom evolution went a little faster, there are those for whom a little slower. Intuitively, I suspect that the value obtained will correlate very well with the length of each specific branch (from the root) for each specific species.
This is what I called the interpretation of Darwinian evolution. But let me specially note that although all Darwinists (that is, classical taxonomists and phylogenetics) should deal with this, they build trees using measures that are more similar to interpreting "many-species origin", and of course it is difficult for them to talk about "antiquity of the species" »By definition, such an interpretation - as mentioned above, there is no direction of evolution and there can be no.
But the opponent turned out to be wrong in his assessment “I think that there will not be much difference between the different types” - it is and significant, and this will be demonstrated further - just look at the resulting tree of evolution.
№5. Method of restoring the direction of evolution
From here can read those who are squeamish about the pathetic text of the amateur that is above.
To understand it is necessary to read the article Systematics of prokaryotes - distant relatives , it describes the basics, which are the input data. Explaining further, I assume that you understand what this graph means, for example, and how it was constructed:
Now we need to figure out how to transform it into a tree with directional evolution, for example:
In this tree we restore the ancestors of the modern genera of bacteria. Modern sorts of bacteria have names and are represented as leaves of a tree, while their ancestors are indicated by a set of numbers.
Each digit is the identifier of the tRNA group that an ancestor must possess in order to pass on to its descendants in the next generation. If he did not possess such a group of tRNA, then we definitely could not get the current state of the relationship (coincidence of identical tRNA), which is available in the column of "multiple origin" above.
Thus, the algorithm for constructing such a tree consists of two parts:
1. Distribution of tRNA into groups, so that the entire analyzed set can be appealed only in groups without switching to single tRNA — this is necessary for two purposes (1) it is an order of magnitude more convenient to deal with groups than with a large set of tRNA. Duplicate information is eliminated, and the group is the minimum unit of divergence. (2) Groups can be sorted by the number of tRNAs entering there. The probability of divergence (separation) of a larger group in different genera is higher with a smaller number of ancestral divergences (branch length).
2. Actually building ancestral tree.
Further, I will describe only the general principle of the implementation of these two parts.
Division into groups:
1. At the entrance there is information of the form:
1 10 000913,003420,00686818,011215,013800,016316,017374,
2 9 000434,000487,005891,005892,011142,01111163,
2 10 000913,003420,006868,007509,011215,013800,016316,017374,
2 8 000487,003420,005891,006678,011163,013218,007509,
it describes a graph of “multi-species origin”, namely a set of links, where “1” is the identification of one kind, “10” is the identification of the second kind, “000913,003420,00686818.011215.013800,016316.017374,” are those tRNAs that identical in both the first and second kind.
2. The first group is created, as a set of all generally different tRNAs.
3. Distribution into groups takes place, if tRNA on relationships between genera belongs to a group, this set is replaced with a group identification, but if the occurrence is partial, then what tRNA is labeled is missing, or, conversely, which tRNA, are only from this group.
4. The division of the group into two. The above made distribution into groups is analyzed, the first partial entry is taken - a new group is created, and the missing part remains with the previous group.
5. Repeating paragraph 3. So gradually, there will be a division into groups without partial occurrences.
6. Groups are sorted by value 1 - a group is, say, a set of 20 tRNAs, and after 300 groups there is an entry of 1-2 tRNA
Ancestral tree construction:
1. Based on the division of the genus links into groups of tRNA, it is possible to recover which groups of tRNA are in each genus. So if between births there is such a connection
1 10 307 | 864 | 867 | 897 | 909 | 911 |
6 10 307 | 862 | 864 | 867 | 897 | 909 | 911 |
This means that groups 307 | 864 | 867 | 897 | 909 | 911 | There is also a 1st kind and a 10th. But the 862 group, for example, has only the 10th and 6th, but not the 1st.
2. All genera do leaves of the tree
3. We take the 1st group (remember that it is the largest, which means it is less fragmented and is younger). We find the smallest common ancestor for all genera that possess this group of tRNA. If there is no such ancestor, create it. If there is, but the smallest common ancestor is not marked with the corresponding identifier of the tRNA group - we label.
4. Repeat p.3. for all groups
Well, the actual result of the tree of the evolution of bacteria can be seen in the picture :
We look in high resolution
PS I understand that I did not provide the actual results of the division into groups, that this tRNA cannot be concretely understood, and the method is described only from a bird's eye view. Really interested, I can provide all the information, but I expect from them that they will try to double-check me at least in something and do not hesitate to make it publicly.
Source: https://habr.com/ru/post/164763/
All Articles