RNAInSpace development, CRA algorithm, Linux code problems and other
I didn’t write for a long time, so I decided to write an interim article on the development of RNAInSpace. The first stage of the articles is compiled in. The folding trajectory of a viroid ribozyme or news from fronts using the RNAInSpace software was obtained . Let's try to start the second stage.
The second stage I was going to start with folding tRNA. There were some problems. On the other hand, there is an interesting CRA algorithm that should help me solve these problems. It is complicated and I do not understand it. But it is implemented in some software mainly for Linux. What is a big fi. In general, everything in order.
PS I am looking for those who understand mathematics and will be able to help me deal with the CRA algorithm. On the other hand, I need the help of those who used Gromacs.
')
I was asked to take the already known tRNA structure so that the model structure could be compared with the real one. I must say, the results are not yet full. But at the same time, I’m wondering if anyone does a simulation on another well-known software, and will show results. In my opinion, there should not be any results, it seems to me that not one known software will not convince tRNA correctly enough.
We begin to understand the secondary structure (see figure), which is supplemented by intercoiled non-canonical hydrogen bonds (they are based on the literature, and measurements of bonds in the .pdb file, which contains the real structure of this tRNA). In my approach, it is argued that if it is possible to form these hydrogen bonds, and some other relationships, the interaction stacking (which is essentially only needed at the intermediate folding stage), then we will get a model structure that is quite close to real.
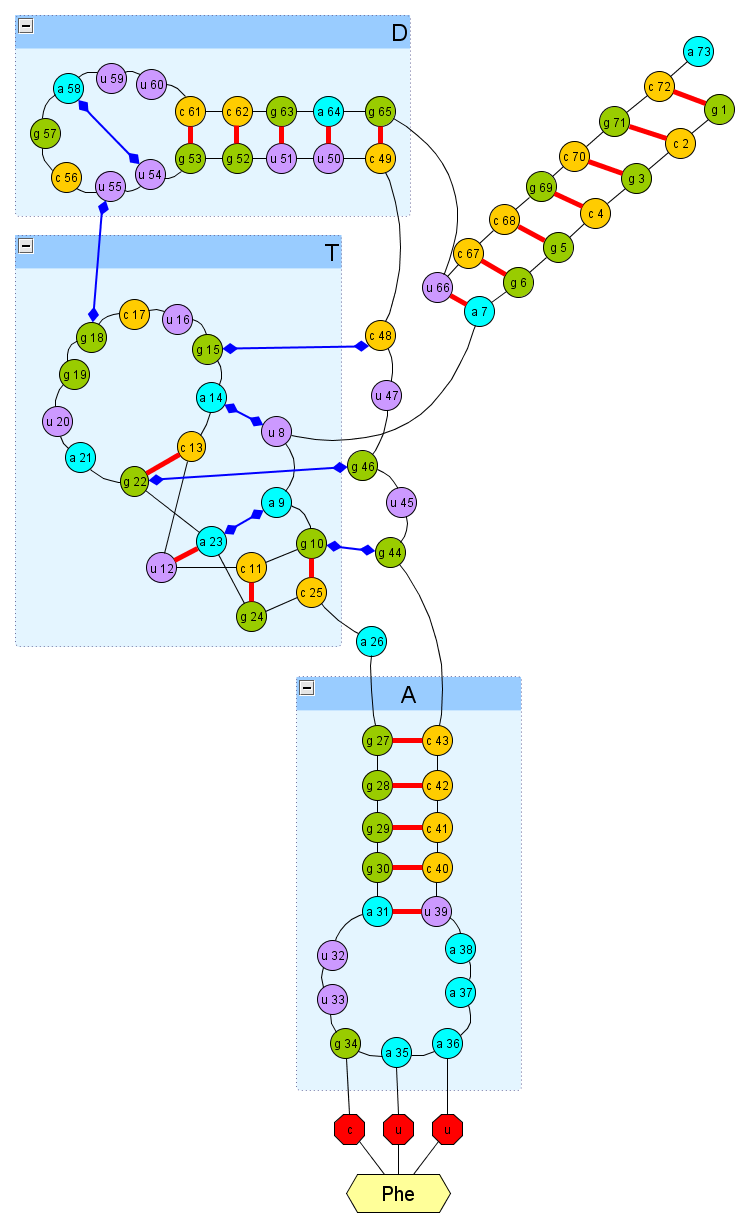
It can be seen that tRNA is much more complicated than in the previous article with a ribozyme. If in a ribozyme it was relatively difficult to form links between three nucleotides, for correct joining of two helices, then for tRNA it is necessary that the T-helix clearly be twisted with nucleotides 44-48, on the other hand, 8-9, and then correctly joined to the helix D .
I will not go into details, but the sequence of folding is not simple here, and so-called contradicts. hierarchical folding models, according to which spirals (secondary structures) are first formed, and then they are joined more closely. This is not true. If the spirals are already formed, then the possibility of docking becomes almost impossible.
At the moment, I can show a model where all spirals are formed, except the final one (1-7 + 66-72). But the joining of the spirals T and D, if necessary, to create all the hydrogen bonds is not yet fully happening.
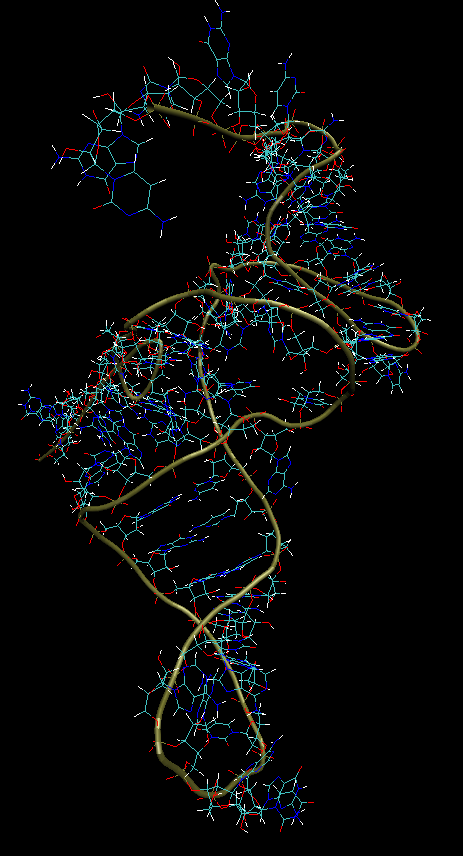
It can be seen that if it were possible to dock properly, and twist the remaining helix, then the tRNA profile would correspond to the real one. And already by how much, measuring by RMSD - this is essentially a secondary issue. But this is exactly how modern scientists in this field compare. Although, while I was studying the real .pdb model of this RNA, I also found several errors in it. Therefore, it is also not perfect, although it was obtained by biological methods. Therefore, measuring by RMSD with a model is to be trusted carefully.
Why is he?
You can, of course, give a few more months and achieve the docking of T and D spirals semi-automatically folding with the help of my RNAInSpace software, interfering where a person sees that folding is wrong, and playing with the sequence of folding and vibration parameters.
But so we can not automate. Of course, folding into a separate helix is now automated for me, but when the helix joins, one helix must fit the other in a completely unique way for a particular nucleotide sequence. These spirals should get used to each other.
And then there is a problem that I felt for a long time, but could not articulate quite clearly. Here are some excerpts and a link:
In general, surprisingly, such an algorithm was not developed either in the work technician or in other areas related to kinematics.
I set out to search and find how the problem I need is solved. It turns out there are such algorithms, and have been developed for a long time, although they have been practically used recently. In Russian, as is customary :) nothing even comes close ... which is a little offensive.
What does he specifically do?
Such algorithms are called concerted rotation algorithm. I came across an article about the so-called. CRISP algorithm, which is made in order to improve the CRA algorithm. The implementation of CRISP and CRA was done in the PHAISTOS software . .
The main chain of RNA (and proteins) consists of atoms linked by bonds. The length of the bonds cannot be changed, but you can only twist the atoms. Accordingly, if we twist an atom, the whole chain is forced to rotate after this atom ( on the folding path of “my” ribozyme, it is clear that one end is fixed and the other moves. The task is to fix both ends at the right points).
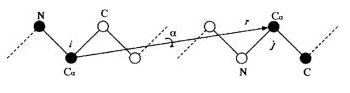
But when folding RNA, it is often necessary to fix the atoms from two sides and be able to rotate only the internal gap. This is what I previously called the "one fundamental problem." These algorithms allow you to do this.
This is done in two stages. First - rotate as we want our atom. If we do not rotate the remaining end, which is fixed, it will break the chain. Well, we go for it. But the second stage, the rotation of the subsequent atom, we must calculate so that the gap disappears completely. As I understand it, there is some kind of inaccuracy, and problems with the fact that such a closure of the gap can lead to the intersection of atoms (bonds) - and therefore different algorithms with different efficiency do it.
How is he doing?
But this is the problem. Maybe there are mathematicians who will help me here, I looked at the formulas, I was horrified and thought to spit. At least, until someone helps. But for not having my knowledge and mathematicians, and understanding the importance of this algorithm, I went to look at ready-made implementations.
Implementation issues
The first problem is that they are written under Linux. The good news is that they are written in C. In general, while looking at the code written under Linux, I can’t overcome the impression that I am returning to the prehistoric era. Talking about compatibility with Windows (that is, supposedly multiplatform) is absolutely not necessary (and I at least 3 “watched” large projects).
I do not even understand why people want to live 30 years in the past. But now is not about that.
Secondly, the quality of the code - Phaistos and Gromacs (about which after) is not just bad, but terrible. Bad, this is when there are no classes and objects, or they are intertwined with the structural style :), horror - when what is written is so that everything is intertwined.
In short, it is difficult to find separately implemented methods, you have to watch the whole package in order to understand something. And these are the whole projects on molecular modeling.
I tried to install Phaistos on Windows ... impossible. It uses the boost library , where I came to an unequivocal conclusion after a couple of days of torment, the library itself is rubbish (using templates, in such an amount that the code is terrible), and it does not compile under Windows in MS Visual Studio. Forgot.
Next, I was lucky to come across another implementation of the CRA method. But also for a healthy project Gromacs. It can be parted in Windows, and it almost works. Only there it is necessary to deal with the tricks of using Gromacs. Maybe there are people who worked with him?
Then maybe the process will go faster. For good I want to make the implementation of the CRA method under MS Visual Studio, as a separate library, at the input you submit the coordinates of the atoms, fixed atoms, the desired rotation and - oplya, the algorithm closes the gap.
The second stage I was going to start with folding tRNA. There were some problems. On the other hand, there is an interesting CRA algorithm that should help me solve these problems. It is complicated and I do not understand it. But it is implemented in some software mainly for Linux. What is a big fi. In general, everything in order.
PS I am looking for those who understand mathematics and will be able to help me deal with the CRA algorithm. On the other hand, I need the help of those who used Gromacs.
')
Folding tRNA
I was asked to take the already known tRNA structure so that the model structure could be compared with the real one. I must say, the results are not yet full. But at the same time, I’m wondering if anyone does a simulation on another well-known software, and will show results. In my opinion, there should not be any results, it seems to me that not one known software will not convince tRNA correctly enough.
We begin to understand the secondary structure (see figure), which is supplemented by intercoiled non-canonical hydrogen bonds (they are based on the literature, and measurements of bonds in the .pdb file, which contains the real structure of this tRNA). In my approach, it is argued that if it is possible to form these hydrogen bonds, and some other relationships, the interaction stacking (which is essentially only needed at the intermediate folding stage), then we will get a model structure that is quite close to real.
It can be seen that tRNA is much more complicated than in the previous article with a ribozyme. If in a ribozyme it was relatively difficult to form links between three nucleotides, for correct joining of two helices, then for tRNA it is necessary that the T-helix clearly be twisted with nucleotides 44-48, on the other hand, 8-9, and then correctly joined to the helix D .
I will not go into details, but the sequence of folding is not simple here, and so-called contradicts. hierarchical folding models, according to which spirals (secondary structures) are first formed, and then they are joined more closely. This is not true. If the spirals are already formed, then the possibility of docking becomes almost impossible.
At the moment, I can show a model where all spirals are formed, except the final one (1-7 + 66-72). But the joining of the spirals T and D, if necessary, to create all the hydrogen bonds is not yet fully happening.
It can be seen that if it were possible to dock properly, and twist the remaining helix, then the tRNA profile would correspond to the real one. And already by how much, measuring by RMSD - this is essentially a secondary issue. But this is exactly how modern scientists in this field compare. Although, while I was studying the real .pdb model of this RNA, I also found several errors in it. Therefore, it is also not perfect, although it was obtained by biological methods. Therefore, measuring by RMSD with a model is to be trusted carefully.
CRA Method
Why is he?
You can, of course, give a few more months and achieve the docking of T and D spirals semi-automatically folding with the help of my RNAInSpace software, interfering where a person sees that folding is wrong, and playing with the sequence of folding and vibration parameters.
But so we can not automate. Of course, folding into a separate helix is now automated for me, but when the helix joins, one helix must fit the other in a completely unique way for a particular nucleotide sequence. These spirals should get used to each other.
And then there is a problem that I felt for a long time, but could not articulate quite clearly. Here are some excerpts and a link:
One fundamental problem is the first strokes to the description of the problem of “teaching two teachers” in this task. We also discussed the possibility of using “inverse kinematics” (which is used to calculate joints in robotics), but all this is terribly slow. Accordingly, the developed method can be applied not only to the problem of folding RNA, but also to improve the solution of problems from the field of robotics. By the way, folding is akin to solving the problem of “parking a car” in every single case of contact between two nucleotides.
In general, surprisingly, such an algorithm was not developed either in the work technician or in other areas related to kinematics.
I set out to search and find how the problem I need is solved. It turns out there are such algorithms, and have been developed for a long time, although they have been practically used recently. In Russian, as is customary :) nothing even comes close ... which is a little offensive.
What does he specifically do?
Such algorithms are called concerted rotation algorithm. I came across an article about the so-called. CRISP algorithm, which is made in order to improve the CRA algorithm. The implementation of CRISP and CRA was done in the PHAISTOS software . .
The main chain of RNA (and proteins) consists of atoms linked by bonds. The length of the bonds cannot be changed, but you can only twist the atoms. Accordingly, if we twist an atom, the whole chain is forced to rotate after this atom ( on the folding path of “my” ribozyme, it is clear that one end is fixed and the other moves. The task is to fix both ends at the right points).
But when folding RNA, it is often necessary to fix the atoms from two sides and be able to rotate only the internal gap. This is what I previously called the "one fundamental problem." These algorithms allow you to do this.
This is done in two stages. First - rotate as we want our atom. If we do not rotate the remaining end, which is fixed, it will break the chain. Well, we go for it. But the second stage, the rotation of the subsequent atom, we must calculate so that the gap disappears completely. As I understand it, there is some kind of inaccuracy, and problems with the fact that such a closure of the gap can lead to the intersection of atoms (bonds) - and therefore different algorithms with different efficiency do it.
How is he doing?
But this is the problem. Maybe there are mathematicians who will help me here, I looked at the formulas, I was horrified and thought to spit. At least, until someone helps. But for not having my knowledge and mathematicians, and understanding the importance of this algorithm, I went to look at ready-made implementations.
Implementation issues
The first problem is that they are written under Linux. The good news is that they are written in C. In general, while looking at the code written under Linux, I can’t overcome the impression that I am returning to the prehistoric era. Talking about compatibility with Windows (that is, supposedly multiplatform) is absolutely not necessary (and I at least 3 “watched” large projects).
I do not even understand why people want to live 30 years in the past. But now is not about that.
Secondly, the quality of the code - Phaistos and Gromacs (about which after) is not just bad, but terrible. Bad, this is when there are no classes and objects, or they are intertwined with the structural style :), horror - when what is written is so that everything is intertwined.
In short, it is difficult to find separately implemented methods, you have to watch the whole package in order to understand something. And these are the whole projects on molecular modeling.
I tried to install Phaistos on Windows ... impossible. It uses the boost library , where I came to an unequivocal conclusion after a couple of days of torment, the library itself is rubbish (using templates, in such an amount that the code is terrible), and it does not compile under Windows in MS Visual Studio. Forgot.
Next, I was lucky to come across another implementation of the CRA method. But also for a healthy project Gromacs. It can be parted in Windows, and it almost works. Only there it is necessary to deal with the tricks of using Gromacs. Maybe there are people who worked with him?
Then maybe the process will go faster. For good I want to make the implementation of the CRA method under MS Visual Studio, as a separate library, at the input you submit the coordinates of the atoms, fixed atoms, the desired rotation and - oplya, the algorithm closes the gap.
Source: https://habr.com/ru/post/154521/
All Articles