Biochemistry on folding. Again in plain language about the resulting folding model.
I have already written more than 7 articles on one of my approaches (a set of algorithms and problems) to the RNA folding task. There were fewer and fewer people reading with each article, and some who confessed that the brain endured after the second article. The comparative success of the first two articles, as compared with the others, seems to lie in the simplicity of the presentation and not going into the details. Although the last articles made it possible to take the demo of my program and feel the problems themselves - this seems less interesting.
Therefore, I will try here to put into simple language another problem that prevents us from solving this problem. And it seems to me that this problem is connected not only with the approach I have chosen to solve, but rather it is common to the task of folding.
In my RNAInSpace software, I realized the ability to “twist” the RNA helix manually so that the geometry and limitations of such a rotation become clear. But since, according to the previous articles, this software is not very interested, then I will not present the next demo version of this software. And let's talk about what happens with me.
')
In order to keep up a conversation with my readers, I’ll go over a number of important notes.
chupvl already asked Have you tested your solution on a real problem, or are you just analyzing a theory at the moment? There I explained to him like this:
Specific example of folding
And now I want to clearly demonstrate what that RNA looks like which I fold.
Below is the “skeleton” of RNA, which I use as a base (2QUS).

I'm trying to minimize the viroid ribozyme NC_003540, whose tertiary structure is unknown. But it must be somewhat similar to the base. Although the primary sequence is almost 80% different. Below is one of the best options that I managed to get. (not automatically, but semi-automatically - similarly as if playing the game FoldIt only in a different way, I have already said that before full automation - as before the moon)

What differences can we see here?
1. It should be understood that the basic sequence (2QUS) is somewhat longer than in the viroid ribozyme, therefore the ends of the RNA in the viroid ribozyme will not form a long helix, as in the basic analogue.
2. The overall styling is fundamentally the same - that is why the viroid ribozyme is essentially collapsed correctly.
3. But there is at least one difference. The loop L2 (see the section in the pictures on the left below) is different.
3.a. In this loop there is one nucleotide, which must form 4 hydrogen bonds with two other nucleotides in loop L1 (the section in the figures on the right below). It is these connections that hold the ribozyme loops together.
(how clearly it looks and what are the problems that I have already solved - I commented earlier here and here )
3.b. Why is there a difference? In the basic ribozyme, this loop is simply longer than in the viroid ribozyme that I study, and the L2 loop has to change its configuration so that it can dock with the L1 loop. And since this is a rather unnatural position of the L2 loop, the configuration of the helix itself (the one above the left) must also change with it.
4. Finally, after all these problems have been resolved, I have already rubbed my hands, thinking that turning the ends of the ribozyme with a sufficiently correct position of the ribozyme core will not be a problem. But wrong. I did not manage to form the necessary hydrogen bonds between the ends of the helix (in the figure one can see that the ends are not close enough to each other).
Why couldn’t the ribozyme succeed?
I began to compare what the problem is with the structure of RNA folded by me. The ends did not want to converge, because they were hampered by a protrusion connecting the spirals L1 and L2 (center top). And it turned out that literally one or two nucleotides, after this protrusion took not the right position. They were not given much attention in modeling. They did not form any hydrogen bonds — and I allowed them to assume any possible position. And they accepted - random. And naturally, the random position did not allow the ribozyme to reach the bottom. (branch: just think how it can, something will turn out during modeling, if in the majority of the methods now used the nucleotides are just placed in a random position, and the correct indicators are selected for some average for the entire RNA molecule indicators?)
I tried to change this position in the already collapsed structure (second figure). But it turned out to be impossible - you need to rebuild almost all the links, i.e destroy all those hydrogen bonds that have already been formed.
And then I thought. Recalled a comment from Wott : the initial state dictates the result , and earlier
This is a fairly correct remark, and for me it was known, earlier in one of my scientific articles I wrote:
Therefore, then I answered in general that such an approach contradicts my results, and that it would be better to start folding from the initial state of a stretched one into a chain than from a half-rolled one.
But there are also so-called interaction stacking: this is when the nucleotides, which simply can be called hexagons, are arranged so that they form a sort of stack of coins. Below in the figure, the first 6 nucleotides (counting from below) are in the stacking interaction.
So, if RNA appears gradually, then before the first hydrogen bond can form, you need at least twenty nucleotides to appear.
And I used to think that they were just stretched into a chain. But they actually appear, one after another, in time for them to accept the position inherent in stacking. And the chain seems to be drawn out, but not quite by accident. And this is precisely what is important for the initial position.
But there was another nuisance. If we remember what a double strand of DNA looks like, then its length can be very large. And there exactly such interaction stacking. But although RNA tends to do this, it only suffices for very small areas, and here in the third figure you can see how, starting with 7 nucleotides, the RNA chain changes direction.
Some conclusions
Probably not very clear what I was driving all this time. Let's try to figure it out.
1. Stacking turns out to be important as creating an initial position, which helps to ensure that when collapsed in nature, the situation did not work out like mine, in the simulation described above, when RNA almost collapsed - but a couple of nucleotides remained in a random position (not related by stacking) - and this prevents further folding.
2. RNA stacking is not as stable as in DNA, where a strict helix is formed. In RNA, at some nodal points, stacking forces act between a pair of nucleotides following each other in nucleotides. And then the chain changes direction.
3. Where the direction of the chain changes, and then when there are complementary pairs of nucleotides, it is then that hydrogen bonds begin to form, which have a stronger stabilizing effect than stacking.
4. But when the chain grows, and is divided into two and more spirals - the loops of these spirals are interconnected by non-standard hydrogen bonds. This is even stronger interaction, and at this time the previous stacking, the approximation of hydrogen bonds, and the shape starting from the loops of the spirals may collapse.
This is the hypothesis - the folding model that I have obtained at the moment. It still needs to be checked finally, but at least many other variations that cannot be fair (such as a hierarchical model (where it is said first about creating secondary structures, and only then combining them into a tertiary one) and others) are now checked.
As a result, it turns out that the task of stacking is to push to the formation of the necessary hydrogen bonds. And without it, hydrogen bonds themselves are not formed.
PS In fact, everything is a little more complicated, but I do not want to ship you. And so it seems just promised, but it turned out not quite. But I am ready to answer any questions where something is not clear.
Therefore, I will try here to put into simple language another problem that prevents us from solving this problem. And it seems to me that this problem is connected not only with the approach I have chosen to solve, but rather it is common to the task of folding.
In my RNAInSpace software, I realized the ability to “twist” the RNA helix manually so that the geometry and limitations of such a rotation become clear. But since, according to the previous articles, this software is not very interested, then I will not present the next demo version of this software. And let's talk about what happens with me.
')
In order to keep up a conversation with my readers, I’ll go over a number of important notes.
chupvl already asked Have you tested your solution on a real problem, or are you just analyzing a theory at the moment? There I explained to him like this:
I fold the ribozyme. Ribozymes have the same basic structure, more precisely, because of what they are distinguished in one class. For comparison, I use another 2QUS ribozyme that is actually available. It is then important for me to see what is the difference and what is the similarity.
Specific example of folding
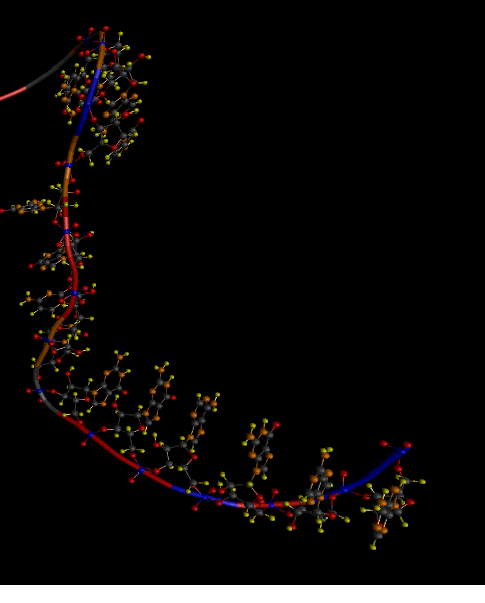
And now I want to clearly demonstrate what that RNA looks like which I fold.
Below is the “skeleton” of RNA, which I use as a base (2QUS).
I'm trying to minimize the viroid ribozyme NC_003540, whose tertiary structure is unknown. But it must be somewhat similar to the base. Although the primary sequence is almost 80% different. Below is one of the best options that I managed to get. (not automatically, but semi-automatically - similarly as if playing the game FoldIt only in a different way, I have already said that before full automation - as before the moon)
What differences can we see here?
1. It should be understood that the basic sequence (2QUS) is somewhat longer than in the viroid ribozyme, therefore the ends of the RNA in the viroid ribozyme will not form a long helix, as in the basic analogue.
2. The overall styling is fundamentally the same - that is why the viroid ribozyme is essentially collapsed correctly.
3. But there is at least one difference. The loop L2 (see the section in the pictures on the left below) is different.
3.a. In this loop there is one nucleotide, which must form 4 hydrogen bonds with two other nucleotides in loop L1 (the section in the figures on the right below). It is these connections that hold the ribozyme loops together.
(how clearly it looks and what are the problems that I have already solved - I commented earlier here and here )
3.b. Why is there a difference? In the basic ribozyme, this loop is simply longer than in the viroid ribozyme that I study, and the L2 loop has to change its configuration so that it can dock with the L1 loop. And since this is a rather unnatural position of the L2 loop, the configuration of the helix itself (the one above the left) must also change with it.
4. Finally, after all these problems have been resolved, I have already rubbed my hands, thinking that turning the ends of the ribozyme with a sufficiently correct position of the ribozyme core will not be a problem. But wrong. I did not manage to form the necessary hydrogen bonds between the ends of the helix (in the figure one can see that the ends are not close enough to each other).
Why couldn’t the ribozyme succeed?
I began to compare what the problem is with the structure of RNA folded by me. The ends did not want to converge, because they were hampered by a protrusion connecting the spirals L1 and L2 (center top). And it turned out that literally one or two nucleotides, after this protrusion took not the right position. They were not given much attention in modeling. They did not form any hydrogen bonds — and I allowed them to assume any possible position. And they accepted - random. And naturally, the random position did not allow the ribozyme to reach the bottom. (branch: just think how it can, something will turn out during modeling, if in the majority of the methods now used the nucleotides are just placed in a random position, and the correct indicators are selected for some average for the entire RNA molecule indicators?)
I tried to change this position in the already collapsed structure (second figure). But it turned out to be impossible - you need to rebuild almost all the links, i.e destroy all those hydrogen bonds that have already been formed.
And then I thought. Recalled a comment from Wott : the initial state dictates the result , and earlier
And the initial position itself dictates what kind of “final” state will be achieved. And the initial state in this case is the process of creating the chain itself. That is, for a good task, it is necessary to solve it iteratively - they added the first, added the following - brought the system to a minimum, added the following - again brought to a minimum.
...
In the same way, it is harmful to consider an extended chain as a whole - this is an implausible state from which it is possible to reach both plausible and not very states, but due to the large number of options, the second will be much larger.
This is a fairly correct remark, and for me it was known, earlier in one of my scientific articles I wrote:
...
In the terminology of game theory, this means that it is necessary to ensure that a given final state can be reached from any initial position of the game. And if, for example, in a game of chess, we know how the game begins and what is the condition of winning, and we know that the rules of chess ensured getting from the beginning of the game to the end, then here, when modeling the folding of macromolecules, we still need to find such rules and prove that they provide a folding process.
Therefore, then I answered in general that such an approach contradicts my results, and that it would be better to start folding from the initial state of a stretched one into a chain than from a half-rolled one.
But there are also so-called interaction stacking: this is when the nucleotides, which simply can be called hexagons, are arranged so that they form a sort of stack of coins. Below in the figure, the first 6 nucleotides (counting from below) are in the stacking interaction.
So, if RNA appears gradually, then before the first hydrogen bond can form, you need at least twenty nucleotides to appear.
And I used to think that they were just stretched into a chain. But they actually appear, one after another, in time for them to accept the position inherent in stacking. And the chain seems to be drawn out, but not quite by accident. And this is precisely what is important for the initial position.
But there was another nuisance. If we remember what a double strand of DNA looks like, then its length can be very large. And there exactly such interaction stacking. But although RNA tends to do this, it only suffices for very small areas, and here in the third figure you can see how, starting with 7 nucleotides, the RNA chain changes direction.
Some conclusions
Probably not very clear what I was driving all this time. Let's try to figure it out.
1. Stacking turns out to be important as creating an initial position, which helps to ensure that when collapsed in nature, the situation did not work out like mine, in the simulation described above, when RNA almost collapsed - but a couple of nucleotides remained in a random position (not related by stacking) - and this prevents further folding.
2. RNA stacking is not as stable as in DNA, where a strict helix is formed. In RNA, at some nodal points, stacking forces act between a pair of nucleotides following each other in nucleotides. And then the chain changes direction.
3. Where the direction of the chain changes, and then when there are complementary pairs of nucleotides, it is then that hydrogen bonds begin to form, which have a stronger stabilizing effect than stacking.
4. But when the chain grows, and is divided into two and more spirals - the loops of these spirals are interconnected by non-standard hydrogen bonds. This is even stronger interaction, and at this time the previous stacking, the approximation of hydrogen bonds, and the shape starting from the loops of the spirals may collapse.
This is the hypothesis - the folding model that I have obtained at the moment. It still needs to be checked finally, but at least many other variations that cannot be fair (such as a hierarchical model (where it is said first about creating secondary structures, and only then combining them into a tertiary one) and others) are now checked.
As a result, it turns out that the task of stacking is to push to the formation of the necessary hydrogen bonds. And without it, hydrogen bonds themselves are not formed.
PS In fact, everything is a little more complicated, but I do not want to ship you. And so it seems just promised, but it turned out not quite. But I am ready to answer any questions where something is not clear.
Source: https://habr.com/ru/post/145917/
All Articles