Part number 2. Introduction to folding biochemistry. Mat. criteria
This is a continuation of the article Part number 1. Introduction to folding biochemistry. From proteins to RNA . Here we describe the covalent and hydrogen bonds mathematically. Let's see what angles we will rotate at the RNA for folding. And we touch the question "what's the difficulty then?"
First two slides:
')
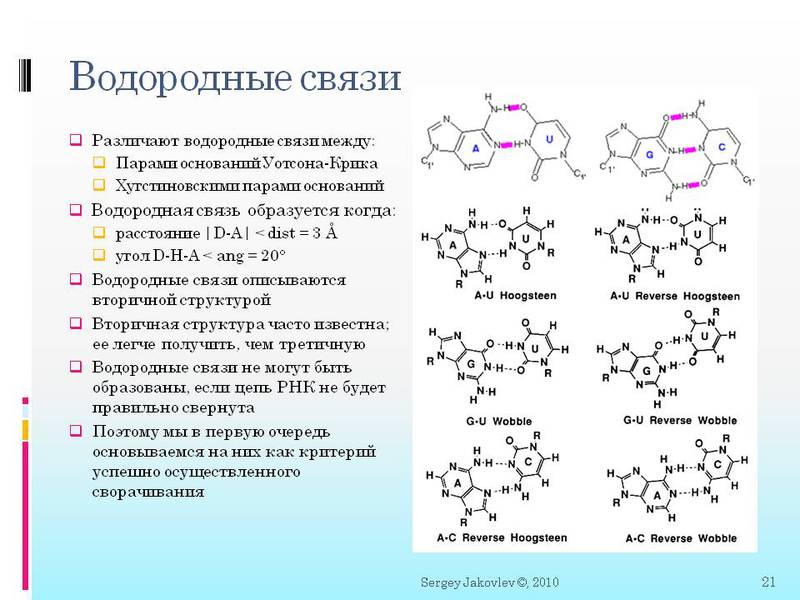

here you see a stricter definition of covalent and hydrogen bonds. And how they are described mathematically. Further it will be important to us:
1. To calculate whether forbidden covalent bonds appeared longer than to actually turn and calculate a dozen hydrogen bonds. Therefore, this is done only when, according to all other criteria, we are satisfied with the state found. Consider that there is just a GetFullCovalentBond () function that tells us if there is at least one forbidden covalent bond. If there is, let's go count further and ignore such states. Although comparatively in the FoldIt game, collisions are rather quickly calculated and get rid of these states from “bad” states. It makes some sense. But alas, I do not have an algorithm for how to do it normally, in general, so far without it.
2. It is important that between the molecules is formed immediately or two or three (depending on the type) of hydrogen bonds. Further we will speak in the context of “between nucleotides formed a hydrogen bond” - means that all two / three bonds were formed.
3. At the beginning, I was very bothered that there could be non-standard hydrogen bonds. Those. not like in DNA only G with C and A with T (and in RNA with U). There may be different surprises. For example, G c U, as well as triple bonds A c with two U at the same time.
4. I will say it again, since this is important - a hydrogen bond is formed when certain atoms are at a distance of <3 angstroms from each other and at a certain angle <20 degrees. All atomic coordinates are calculated in angstroms. And there is a ScoreGroup function (int N1, int N2), where two numbers of nucleotides (molecules) in the chain are fed, and we get the answer to how close they are to create all the necessary hydrogen bonds. It is important here that the answer is not just yes / no, but as a function of approaching “yes”. What exactly is the function will be discussed in subsequent articles.
Now how can we turn the chain. Let's start with turning only one nucleotide:

The chain turns only by turning 9 torsion angles. No way else she can not move - is prohibited by biology (energy). 6 corners are located in the so-called main circuit, what is shown in the last article in a solid line. Here are indicated in Greek letters starting with alpha. Three more angles in the side chain are strongly dependent on each other, i.e. you cannot rotate them independently - so we will call this next the number 7 angle - but in fact there will be a combination of three corners. A side chain is a radical, i.e. what nucleotides a, u, c, g actually differ in - and the main chain (12 atoms out of 28-33) is the same.
If we have only one nucleotide - it is clear that in 6 corners you can rotate almost without restrictions. Yes, and 7 th very few bans. Nothing interferes. If there is already a chain of 2 nucleotides, then prohibitions appear - atoms of one nucleotide can jump on the atoms of another. But since the bundle is not big (take the beads - the two balls standing next to it are not strong, they can collide with them during rotation - the length of the string is small between them) - then there are not too many restrictions. But if you take 6 or more bans can be a lot.
And then a turn by one angle of 0.1 degrees can allow it to turn on a different angle, for example, to form a hydrogen bond, and without this it will not work.
What is the difficulty? Calculate all the combinations, say for a day, only for a chain of 2-3 nucleotides, 4 already needs a week-month, and then years :)
But not all corners are correlated with each other. There are those who have little influence on each other. But no one knows what it is in this case.
But it’s not possible to take and set the required angles, so I want to develop an iterative procedure in order to get all the best and the best condition step by step. But I did this experiment. Took only 3 nucleotides (biologically degenerate state). And he made turns at the beginning of the first nucleotide, found the best condition, then fixing the second best, then the third one (one iteration - miscalculation let's say 1500 + 1500 + 1500 turns) fix the best ... and on and on. The algorithm eventually finds such a state with a score of -9.41.
If we do this, we turn the first nucleotide for 1500 turns, then for each of the 1500 options we turn the second one by 1500, then for the first-second combination (1500 * 1500 options) we turn the third one by 1500 turn options. We get a really complete bust of about 3 billion options, and we get the best state -13.87.
But the way of the most complete search is closed, but I would like to reduce it to the variant of local approximation (the first option). How to do this we will talk in the next part.
For an in-depth understanding I will give two links:
1. About the details of the experiment described above.
2. On the freedom of nucleotide oscillations in the RNA helix , it is clearly seen that the laws are as non-linear as possible and there is no simple logic.
First two slides:
')
here you see a stricter definition of covalent and hydrogen bonds. And how they are described mathematically. Further it will be important to us:
1. To calculate whether forbidden covalent bonds appeared longer than to actually turn and calculate a dozen hydrogen bonds. Therefore, this is done only when, according to all other criteria, we are satisfied with the state found. Consider that there is just a GetFullCovalentBond () function that tells us if there is at least one forbidden covalent bond. If there is, let's go count further and ignore such states. Although comparatively in the FoldIt game, collisions are rather quickly calculated and get rid of these states from “bad” states. It makes some sense. But alas, I do not have an algorithm for how to do it normally, in general, so far without it.
2. It is important that between the molecules is formed immediately or two or three (depending on the type) of hydrogen bonds. Further we will speak in the context of “between nucleotides formed a hydrogen bond” - means that all two / three bonds were formed.
3. At the beginning, I was very bothered that there could be non-standard hydrogen bonds. Those. not like in DNA only G with C and A with T (and in RNA with U). There may be different surprises. For example, G c U, as well as triple bonds A c with two U at the same time.
4. I will say it again, since this is important - a hydrogen bond is formed when certain atoms are at a distance of <3 angstroms from each other and at a certain angle <20 degrees. All atomic coordinates are calculated in angstroms. And there is a ScoreGroup function (int N1, int N2), where two numbers of nucleotides (molecules) in the chain are fed, and we get the answer to how close they are to create all the necessary hydrogen bonds. It is important here that the answer is not just yes / no, but as a function of approaching “yes”. What exactly is the function will be discussed in subsequent articles.
Now how can we turn the chain. Let's start with turning only one nucleotide:
The chain turns only by turning 9 torsion angles. No way else she can not move - is prohibited by biology (energy). 6 corners are located in the so-called main circuit, what is shown in the last article in a solid line. Here are indicated in Greek letters starting with alpha. Three more angles in the side chain are strongly dependent on each other, i.e. you cannot rotate them independently - so we will call this next the number 7 angle - but in fact there will be a combination of three corners. A side chain is a radical, i.e. what nucleotides a, u, c, g actually differ in - and the main chain (12 atoms out of 28-33) is the same.
If we have only one nucleotide - it is clear that in 6 corners you can rotate almost without restrictions. Yes, and 7 th very few bans. Nothing interferes. If there is already a chain of 2 nucleotides, then prohibitions appear - atoms of one nucleotide can jump on the atoms of another. But since the bundle is not big (take the beads - the two balls standing next to it are not strong, they can collide with them during rotation - the length of the string is small between them) - then there are not too many restrictions. But if you take 6 or more bans can be a lot.
And then a turn by one angle of 0.1 degrees can allow it to turn on a different angle, for example, to form a hydrogen bond, and without this it will not work.
What is the difficulty? Calculate all the combinations, say for a day, only for a chain of 2-3 nucleotides, 4 already needs a week-month, and then years :)
But not all corners are correlated with each other. There are those who have little influence on each other. But no one knows what it is in this case.
But it’s not possible to take and set the required angles, so I want to develop an iterative procedure in order to get all the best and the best condition step by step. But I did this experiment. Took only 3 nucleotides (biologically degenerate state). And he made turns at the beginning of the first nucleotide, found the best condition, then fixing the second best, then the third one (one iteration - miscalculation let's say 1500 + 1500 + 1500 turns) fix the best ... and on and on. The algorithm eventually finds such a state with a score of -9.41.
If we do this, we turn the first nucleotide for 1500 turns, then for each of the 1500 options we turn the second one by 1500, then for the first-second combination (1500 * 1500 options) we turn the third one by 1500 turn options. We get a really complete bust of about 3 billion options, and we get the best state -13.87.
But the way of the most complete search is closed, but I would like to reduce it to the variant of local approximation (the first option). How to do this we will talk in the next part.
For an in-depth understanding I will give two links:
1. About the details of the experiment described above.
2. On the freedom of nucleotide oscillations in the RNA helix , it is clearly seen that the laws are as non-linear as possible and there is no simple logic.
Source: https://habr.com/ru/post/140154/
All Articles