Part number 1. Introduction to folding biochemistry. From proteins to RNA
At once it must be said that I will pose the question of bio-computation from a certain cybernetic-geometric point of view. This is my name and this direction is not common. I am sure that it will be easier to understand this for those who are not in the subject of this biological perspective. Those who are already in the subject are ready to discuss with you and show why traditional methods are not suitable from the point of view of the cybernetic approach (but in this article you are not my audience - excuse me, but I am sure you will also find it useful as an extension of the world view on the problematic).
Practical application to biologists has more to deal with protein folding. To a certain extent, a lot of practical tasks can be reduced to this task (knowledge of how protein is folded), the main of which is the development of drugs to combat viruses and diseases.
But this problem is not solved in general. It is like unsolved problems in mathematics, only with a biological context (see the Levintal paradox). Biologists can only with a certain error by means of bio-experiments see the state in the already folded state, but it is not yet possible to trace how this happens. But all this is also very expensive. Why they do computer computing — it's cheap, even though thousands of computers are used in distributed projects.
')
But the introduction is enough, then from the ship to the ball ...
First, why RNA, but not protein? Just because it is easier. It is impossible to understand how proteins fold, not understanding how simpler RNA molecules fold. After all, we are not biologists, and we are not concerned with a practical biological result, but with an understanding of the process mathematically / cybernetically.
Here there is such a game FoldIt . And there is a reader question :
I answer. This game is a cry of despair . There are absolutely no algorithms that fold. Even a complete bust does not help. Levintal's paradox shows that the calculation of the folding of a molecule can last longer than the whole life of the Universe.
Players offer such an experiment in the game. Initially, you are given a molecule already somehow folded and with some predefined structure (helix / sheets / loop). If you reset and load again, you will be given a slightly different structure and initial state. Experts already calculate this initial state for you and simply distribute between you (as between computers) what you need to calculate. Do the following. Using the "gum" stretch the protein chain in line. Remove all spirals and sheets from the structure, i.e. turn everything into a loop (actually just a chain without a structure). Before starting these actions, note how many points you were given (state rating). Compare with what happened. We see the difference is not very big. Right? But now try to roll at least to the state that was originally. None of the available tools will help you automatically. If you do not remember how the structure looks like - you do not even have any hypotheses how to do it. I would be glad if as David Baker says
but I'm a pessimist here.
Secondly, biologists like to talk about the minimum energy of the folded protein, and supposedly when the energy is minimal, the protein takes the most stable state. But it is not proven, it is a theory. This energy is affected by so many factors that are simply not possible to calculate.
But we will act completely differently. We will ONLY take into account the importance of the formation of hydrogen bonds and the absence of forbidden covalent bonds (which will explain this later). Why? Because in my experiments, this was largely enough to get results comparable to the results of serious research projects (not quantitatively (because I have only one computer), but qualitatively).
And most importantly, we postulate this basic principle of the cybernetic-geometric approach:
Now a few pictures (by the way, how to adjust the size of the picture, tell me who knows: it is height = "20" width = "20" - but as you can see the size is big):
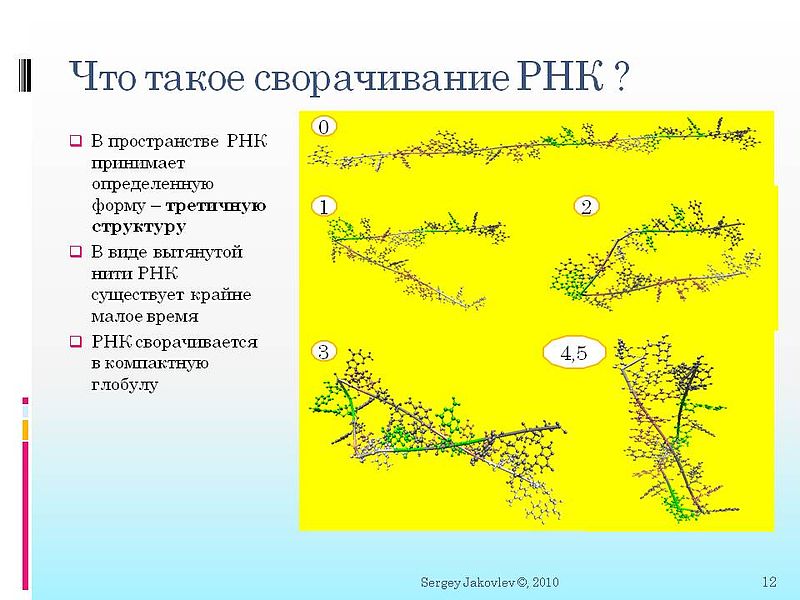
In the picture: step 0. See the RNA chain. Who read my programmatically directed article - this is precisely the initial graph of the circuit in question.
Then you see how the chain in several steps tends to curl up into a spiral. Well constructed spiral in the following figure.

So, I explain what covalent and hydrogen bonds are and for the first time is enough.
In step 0 (first picture) - you can see that the points are connected by lines. Dots are separate atoms. And the lines are allowed covalent bonds. In the process of folding it is impossible to allow any other covalent bonds to form, roughly speaking, that the allowed covalent bonds intersect. In the game FoldIt, this is just shown by a red dogrose and is heavily penalized by points. It’s just that in reality such connections cannot be formed - there is little energy for this, and of course it’s easy to program it so programmatically that it’s easy to push. These are forbidden covalent bonds.
Hydrogen bonds are visible in the second figure red dashed lines between atoms. These bonds are more easily formed and basically they keep RNA in a collapsed state.
In the second part, we will talk about what hydrogen bonds are, how to describe their formation mathematically and the initial steps to what logic to start folding RNA. But I would like feedback - write who understood what and are ready to read the second part . Or I missed something to understand, it will only be more difficult, so speak better right away.
Practical application to biologists has more to deal with protein folding. To a certain extent, a lot of practical tasks can be reduced to this task (knowledge of how protein is folded), the main of which is the development of drugs to combat viruses and diseases.
But this problem is not solved in general. It is like unsolved problems in mathematics, only with a biological context (see the Levintal paradox). Biologists can only with a certain error by means of bio-experiments see the state in the already folded state, but it is not yet possible to trace how this happens. But all this is also very expensive. Why they do computer computing — it's cheap, even though thousands of computers are used in distributed projects.
')
But the introduction is enough, then from the ship to the ball ...
First, why RNA, but not protein? Just because it is easier. It is impossible to understand how proteins fold, not understanding how simpler RNA molecules fold. After all, we are not biologists, and we are not concerned with a practical biological result, but with an understanding of the process mathematically / cybernetically.
Here there is such a game FoldIt . And there is a reader question :
I did not quite understand how manual busting reduces modeling by an order of magnitude. The way people fold their squirrels in general is also by chance, people in fact do not even understand how it should be rolled up. And if we assume that people will simply reject a number of unrealistic options on any grounds, why can't we algorithmize this in folding @ home?
I answer. This game is a cry of despair . There are absolutely no algorithms that fold. Even a complete bust does not help. Levintal's paradox shows that the calculation of the folding of a molecule can last longer than the whole life of the Universe.
Players offer such an experiment in the game. Initially, you are given a molecule already somehow folded and with some predefined structure (helix / sheets / loop). If you reset and load again, you will be given a slightly different structure and initial state. Experts already calculate this initial state for you and simply distribute between you (as between computers) what you need to calculate. Do the following. Using the "gum" stretch the protein chain in line. Remove all spirals and sheets from the structure, i.e. turn everything into a loop (actually just a chain without a structure). Before starting these actions, note how many points you were given (state rating). Compare with what happened. We see the difference is not very big. Right? But now try to roll at least to the state that was originally. None of the available tools will help you automatically. If you do not remember how the structure looks like - you do not even have any hypotheses how to do it. I would be glad if as David Baker says
sincerely believes that somewhere in the world live talents who have an innate ability to calculate in their minds 3D models of proteins. Some 12-year-old boy from Indonesia will see the game and will be able to solve tasks that even a supercomputer cannot do.
but I'm a pessimist here.
Secondly, biologists like to talk about the minimum energy of the folded protein, and supposedly when the energy is minimal, the protein takes the most stable state. But it is not proven, it is a theory. This energy is affected by so many factors that are simply not possible to calculate.
But we will act completely differently. We will ONLY take into account the importance of the formation of hydrogen bonds and the absence of forbidden covalent bonds (which will explain this later). Why? Because in my experiments, this was largely enough to get results comparable to the results of serious research projects (not quantitatively (because I have only one computer), but qualitatively).
And most importantly, we postulate this basic principle of the cybernetic-geometric approach:
we idealize the folding process without considering any other interactions than hydrogen bonds. Thus, in modeling, we intentionally proceed from simplification, idealization, as if answering the questions: “how will the folding process go if RNA only seeks to form hydrogen bonds?” And “what is the“ pure ”contribution of hydrogen bond formation to the folding process? "(This is from my scientific article)
Now a few pictures (by the way, how to adjust the size of the picture, tell me who knows: it is height = "20" width = "20" - but as you can see the size is big):
In the picture: step 0. See the RNA chain. Who read my programmatically directed article - this is precisely the initial graph of the circuit in question.
Then you see how the chain in several steps tends to curl up into a spiral. Well constructed spiral in the following figure.
So, I explain what covalent and hydrogen bonds are and for the first time is enough.
In step 0 (first picture) - you can see that the points are connected by lines. Dots are separate atoms. And the lines are allowed covalent bonds. In the process of folding it is impossible to allow any other covalent bonds to form, roughly speaking, that the allowed covalent bonds intersect. In the game FoldIt, this is just shown by a red dogrose and is heavily penalized by points. It’s just that in reality such connections cannot be formed - there is little energy for this, and of course it’s easy to program it so programmatically that it’s easy to push. These are forbidden covalent bonds.
Hydrogen bonds are visible in the second figure red dashed lines between atoms. These bonds are more easily formed and basically they keep RNA in a collapsed state.
In the second part, we will talk about what hydrogen bonds are, how to describe their formation mathematically and the initial steps to what logic to start folding RNA. But I would like feedback - write who understood what and are ready to read the second part . Or I missed something to understand, it will only be more difficult, so speak better right away.
Source: https://habr.com/ru/post/140150/
All Articles