Practical bioinformatics p.5. DNA sequencing
 In an effort to talk about the most difficult, as quickly as possible, obviously, you forget about the simplest. And, in my case, not only about a simple, but also about an important link. Causation slightly disrupted. In my previous articles ( 1 , 2 , 3 , 4 ) the mathematical aspect and programming are described, but there is practically no biology in them. Therefore, this article is about exactly what part of molecular biology they are trying to uncover, predict, see and solve the programs and algorithms that I describe.
In an effort to talk about the most difficult, as quickly as possible, obviously, you forget about the simplest. And, in my case, not only about a simple, but also about an important link. Causation slightly disrupted. In my previous articles ( 1 , 2 , 3 , 4 ) the mathematical aspect and programming are described, but there is practically no biology in them. Therefore, this article is about exactly what part of molecular biology they are trying to uncover, predict, see and solve the programs and algorithms that I describe.The drawing with the image of an egg cell and sperm cells on the surface symbolizes the stage I missed when everything is just being born. An interesting fact is that the union of two cells gives rise to about 10 trillion cells of the human body.
Let's start with a little excursion into molecular biology. I will try to describe in simple words complex things, omitting the details. DNA is in the cell. We consider exactly the DNA and the processes that take place on it, we are not very interested in exactly where it is located in the cell. It is necessary to clarify that examples will be given for eukaryotic cells with which I work, but most likely many things are suitable for prokaryotic .
')
All processes in the cell begin with DNA, it is on the DNA that the nucleotide sequences are located, the copying of which is later responsible for the reactions and transformations in the cells. We are interested in the copying process, called transcription . The process of copying transcripts and the number of final products obtained is called gene expression. But we call it gene expression and we measure exactly the number of transcript copies (regardless of the number of final products obtained).
Below is an illustration of a textbook on biology, called “the four main genetic processes occurring in a cell” (see Fig. 1). At the moment I am engaged in the processes depicted in this figure between the numbers 1 and 2. The technologies that help to study them are called DNA-seq and RNA-seq. These technologies can be used both individually and jointly. In this article I will focus in more detail on the mechanisms of DNA-seq.
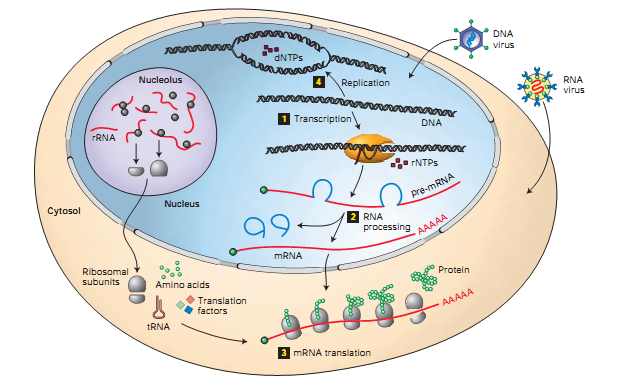
Fig. one
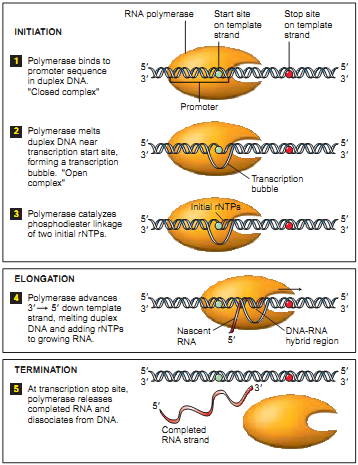 As a special case of DNA-seq, consider the “DNA-seq Pol II” (also see “ Preparing to work with ZINBA ”). The basis of the understanding of the result obtained with its help lies in the process of transcription. An important role in the process of transcription is played by the RNA polymerase protein ( RNA Polymerase ). Polymerase is fixed in a special section of DNA (see Fig. 2.1), which is called a promoter. While the preparatory operations of the transcription process are underway (the initiation stage), the polymerase remains fixed on the promoter. Polymerase begins to create a copy of the upper helix from the start of the site (the beginning of the replicated chain, indicated by a blue dot in the figure). Then, moving at a speed of about 1000 bases per minute (at 37C), it copies the DNA segment to the stop site (the end of the replicated chain, indicated in the figure by a red dot). Once again, the polymerase promoter is in the position of the promoter most of the time, and less in each section of the copied DNA. For further study, scientists are interested in the promoter and start site, which are a necessary condition for the start of transcription. Also in the promoter laid information on the number of copies, which is also an important object of study.
As a special case of DNA-seq, consider the “DNA-seq Pol II” (also see “ Preparing to work with ZINBA ”). The basis of the understanding of the result obtained with its help lies in the process of transcription. An important role in the process of transcription is played by the RNA polymerase protein ( RNA Polymerase ). Polymerase is fixed in a special section of DNA (see Fig. 2.1), which is called a promoter. While the preparatory operations of the transcription process are underway (the initiation stage), the polymerase remains fixed on the promoter. Polymerase begins to create a copy of the upper helix from the start of the site (the beginning of the replicated chain, indicated by a blue dot in the figure). Then, moving at a speed of about 1000 bases per minute (at 37C), it copies the DNA segment to the stop site (the end of the replicated chain, indicated in the figure by a red dot). Once again, the polymerase promoter is in the position of the promoter most of the time, and less in each section of the copied DNA. For further study, scientists are interested in the promoter and start site, which are a necessary condition for the start of transcription. Also in the promoter laid information on the number of copies, which is also an important object of study.The DNA-seq Pol II process with precipitation takes place in several stages: 1. fix the current position of the polymerase chemically or at a temperature (fixation of the polymerase means that the processes have stopped, the polymerase does not move more along the DNA); 2. in an arbitrary way (ultrasound, MNase , etc.) we cut the DNA (in most cases, the sections of DNA protected by polymerase are cut); 3. using immunoprecipitation of chromatin (an antibody that is immune to all other proteins, except Pol II), we select those regions that contain polymerase. As a result, we obtain fragments of about 150 bases in length, which we will later send to the sequencer. It should be emphasized that the cutting of the polymerase does not occur strictly along the edges. Thus, the process turns out to be the following: we prepared millions of the same DNA from different cells, fixed polymerases, most of which are on the promoter, and some are randomly distributed throughout the gene. The result of the digitization of the described process is observed in the next figure, shown in one of the articles (see Fig.3). The green lines are circled, defining the intended promoter (high-density reads), followed by low-density reads, corresponding to polymerase in motion.
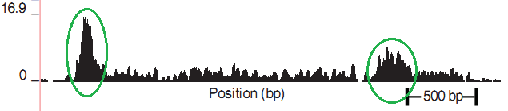
Fig. 3
Next, I will briefly talk about some other proteins. At the top of Figure 4, you can see how the DNA is packaged (see Fig. 4.1.), And at the bottom of the figure shows in more detail the transcription process described above (see Fig. 4.2.). As can be seen in Figure 4.1., The DNA packaging is very dense, the double helix is wound on the nucleosome, the length of the DNA coil is approximately 146 bases. The nucleosome consists of histones. Histones can have various modifications; modification of histones is the subject of a huge separate article. Antibodies are developed for a specific modification of the histone, and it is the DNA segments that are wrapped around this modification of the histone that will be precipitated (filtered). Let us give an example of the designation of the modification: H3K4Me3. It will be read as trimethylation of lysine in the fourth position of the histone H3. Such modifications of histones can be numerous, and all of them are scattered along DNA, therefore, in the article “ Preparing to work with ZINBA ” in Figure 2, which depicts the H3K27Me3 landscape (trimethylation of lysine 27 of histone 3), such frequent accumulation of peaks is observed.
There are DNA-seq technologies without precipitation, when DNA is simply cut, for example, with the help of DNase enzyme, and we get a huge amount of fragments. In most cases, the fragments will correspond to the turns that are wrapped around the nucleosomes, since the DNase cuts between them. The resulting fragments are sent to the sequencer. It is believed that with deep sequencing (a huge number of fragments, about 100 million reads, goes to the sequencer, while it is expensive) by the resulting landscape you can recognize exactly where the protein is. Moreover, the landscape will look like a volcano, a small depression at the top must correspond to a squirrel. Most often, the DNase method is used to find DNA regions sensitive to this enzyme, i.e. those plots that are best cut.
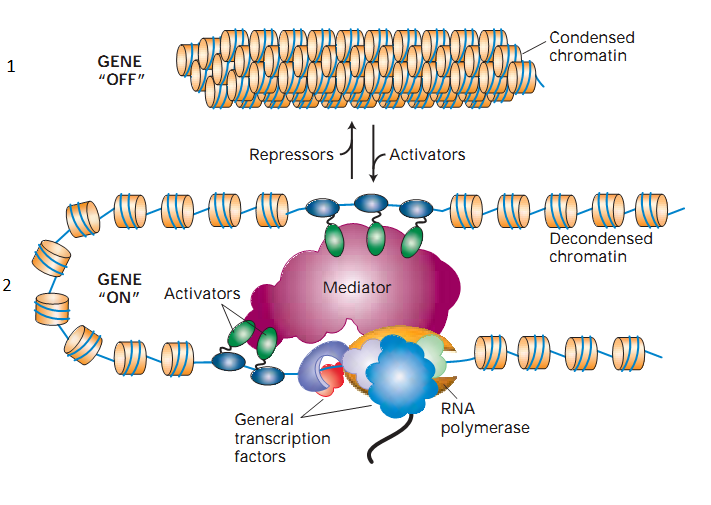
Fig. four
At the bottom of the figure, we see RNA polymerase, as well as multi-colored clouds and ovals placed in it and on DNA. These additional elements are called transcription factors; they play an important role in the regulation of gene expression. Transcription factors can either increase or decrease or simply block the possibility of adding other transcription factors, thereby implicitly leading to the regulation of expression. They are also proteins. As with all other proteins, antibodies are being developed for them. As an example of a transcription factor, take the CTCF protein; one of its roles is to block the operation of other transcription factors. DNA-seq experiments with it are described in the article “ Preparing to work with ZINBA ”, the corresponding CTCF chromatin immunoprecipitation landscape can be seen in the following figure . As can be seen, the regions protected by this protein are small, therefore, the spread in the neighborhood is not high, only 150-200bp.
An exemplary diagram of the types of DNA-seq experiments (sequencing methods):
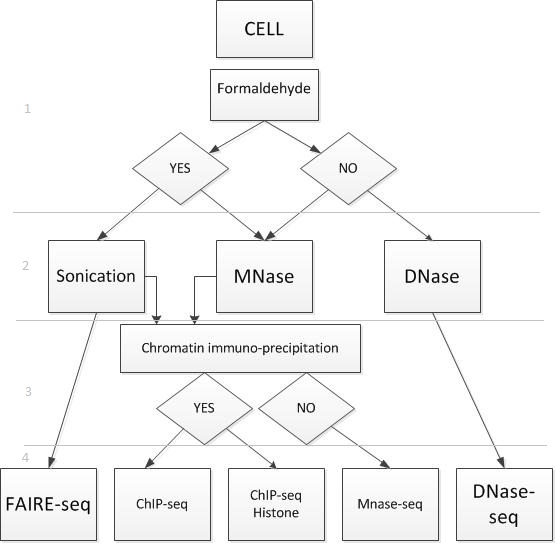
Fig. five
I have broken the scheme into four conditional levels. The first level is fixation, it can be carried out with or without formaldehyde. The second level - the method of cutting DNA. The third level is filtering by size, in addition to this filtering can be carried out using chromatin immunoprecipitation. The fourth level is the sequencing method.
Thus, each type of experiment corresponds to the method of sequencing. Depending on the sequencing method, different kinds of fragments are created. Consider those experiments, the results of which are fragments containing protein. Protein can be placed in the center of the piece or on one of the two sides of the piece. The sequencer does not decode the entire segment, but only a small part of it from the 5 'end of the helix of each fragment. Decoded pieces are called reads. In Figure 6, the red dots mark the 5 'ends of each helix. Blue and red arrows marked the beginning of the reads. These beginnings are randomly distributed from the 5 'end of each helix to the protein border.
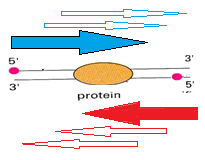
Fig. 6
The experiment involves a lot of the original molecules, their DNA is cut in some neighborhood of the protein, when many reads are mapped onto the coordinate axis (of one of the proteins), a picture similar to this is obtained:

Fig. 7
Reeds from the upper helix (“+” strand) are displayed above the coordinate axis (dark blue), and reads from the lower helix (“-” strand) are displayed below the axis (light blue). To say exactly where the protein was impossible, it is assumed that the protein binding site is located between these two peaks.
The resulting picture resembles a signal, and therefore some algorithms were applied to the data for analyzing signals, one of which is described in this article habrahabr.ru/blogs/algorithm/135281 - “What are hidden Markov models”.
I hope that after this article the subject has become more understandable, and further study will not cause a little horror. If someone is interested in a more detailed presentation of the material, there is a wonderful book Molecular Cell Biology edited by Lodish, in which the basics of molecular biology are very informative and accessible, some of the illustrations are borrowed from it.
Review by Andrey Kartashov, Cincinnati, OH, porter@porter.st.
Source: https://habr.com/ru/post/137626/
All Articles