Practical bioinformatics p.4. Getting ready to work with ZINBA
 In the modern world of data analysis, using only one method or only one approach means that sooner or later you will be confronted with the fact that you were wrong. To analyze the data, various methods are combined, they compare the result and, based on the comparison, they are already making more accurate predictions. ZINBA uses exactly this approach. The developers combined a variety of methods for analyzing DNA-seq experiments in a single package. This package is written for the R statistical software. So what does ZINBA do? It finds various enriched regions even in cases where some of them have been enhanced, for example, chemically or have a different degree of signal-to-noise ratio.
In the modern world of data analysis, using only one method or only one approach means that sooner or later you will be confronted with the fact that you were wrong. To analyze the data, various methods are combined, they compare the result and, based on the comparison, they are already making more accurate predictions. ZINBA uses exactly this approach. The developers combined a variety of methods for analyzing DNA-seq experiments in a single package. This package is written for the R statistical software. So what does ZINBA do? It finds various enriched regions even in cases where some of them have been enhanced, for example, chemically or have a different degree of signal-to-noise ratio.At first I expected to make an overview article about the ZINBA software product, but the more I read about the methods used in it, the deeper I dig into the algorithms and definitions. And when I further summarized my knowledge, I realized the fact that more than one article had already accumulated data, and without introducing it, it would be difficult to touch the subject matter. In this topic, I provide brief excerpts from the articles mentioned in the description of the ZINBA program, complementing them with my own comments. I will wait for your comments to get to the truth together.
Far-reaching plans for regulatory biology are to learn how the genome encodes a variety of gene expression mechanisms. The link between coding and these mechanisms is found due to the possibility of finding protein binding sites throughout the genome using chromatin immunoprecipitation ( ChIP ) and gene expression (RNA-seq). Going for evidence is far from necessary, there is the project Encyclopedia of DNA Elements (ENCODE [1]), within which most of the functional elements of the genome are recognized. The first steps were made with the help of the microchip (“microarray”) method, but deep DNA sequencing (ChIP-seq & DNA-seq, sometimes the term NGS next generation sequencing is found) caught up and overtook this technology.
RNA-seq and ChIP-seq have the following positive differences: they increase the accuracy, specificity, sensitivity of measurements and allow you to view the entire genome at once. In general, RNA-seq and ChIP-seq have some similarities with such a predecessor as a microchip, but particulars differ greatly. But sequencing still suffers from a number of difficulties, although it is much lighter than the microchip in these matters. Due to the fact that the cut DNA fragments are rather short, there is a possibility that the fragments may occur several times in the genome, be parts of the repeatable (repetitive) part of the DNA, or turn out to be common fragments for some start sites.
It is believed that data processing should consider the ChIP reaction as enrichment. Because approximately 60-99% of the fragments in the prepared DNA solution are usually the background, and the remaining 1-40% belong to the fragments, the protein of which could be bound ( cross-link ) with the antibody [2]. It is this smallest part of the solution that will be sequenced. The fact that the level of contamination is high must be taken into account when calculating the enrichment. The number of reads per genome in some cases is incomparably small, for example, for the mammalian genome, the number of reads is less than 1% of the total genome length. The data obtained by sequencing require, obviously, new algorithms and software.
 Fig. one
Fig. oneThis slide schematically displays the hierarchy of ChIP-seq and RNA-seq analysis. Usually the analysis goes through all the stages from the bottom up. Different programs are applied at each level of the scheme and are sometimes divided into programs specifically for ChIP-seq and RNA-seq. The output of one program is the input to the other (pipeline). As can be seen from the diagram, all programs first pass through the definition of reads on the genome (Maps read), with the exception of the assembly of transcripts, the result of which are the alleged transcriptomes [3]. After finding the reads on the genome, the output of such a program is transferred to the input of the program for data collection and identification (Aggregate and identity). For example, to determine enriched regions or read densities on a known annotation. Also, these programs may include some subsequent analysis of both binding sites (for DNA-seq) and new unexplored fragments of transcription-isoformism (for ChIP-seq). Further analysis of a higher level. It includes the definition of a conservative motif or expression level, the result of which could be the discovery of a new gene model. The level of integration (Integrate) shows that previously obtained results can be used for current data processing (not necessarily in the framework of the current experiment, laboratory or country).
So what do we get after the reads were found on the genome? How do these data look and how do they differ from each other? There are sites on DNA (binding sites) to which a particular protein can attach; we will call these sites binding sites or simply sites. For example, a CTCF site means that the CTCF protein can gain a foothold in this region. Scientists have found an antibody to CTCF protein and with the help of antibodies were able to precipitate the corresponding sections of DNA. But, as we already know, arbitrary ones can be attached to specific antibodies along with special sections. The rest of the methods work in a similar way: the protein is fixed on DNA, the antibody is fixed on the protein, only a sample of DNA segments is changing. Thus, the specificity of the selected areas leaves its mark, and the design landscape of each method will have its own: somewhere narrow peaks, as in the CTCF method, the graph of a; somewhere steep peaks with a large adjacent territory, for example, RNA polymerase II method, graph b; during the deposition of the histone modification, a continuous “fringe” is obtained; this is due to the peculiarity of the arrangement of the histones, figure c. On the graphs, the coordinate on the chromosome is plotted along the axis x, and the density of reads along the axis y. The data for the figure are taken from [4].
')
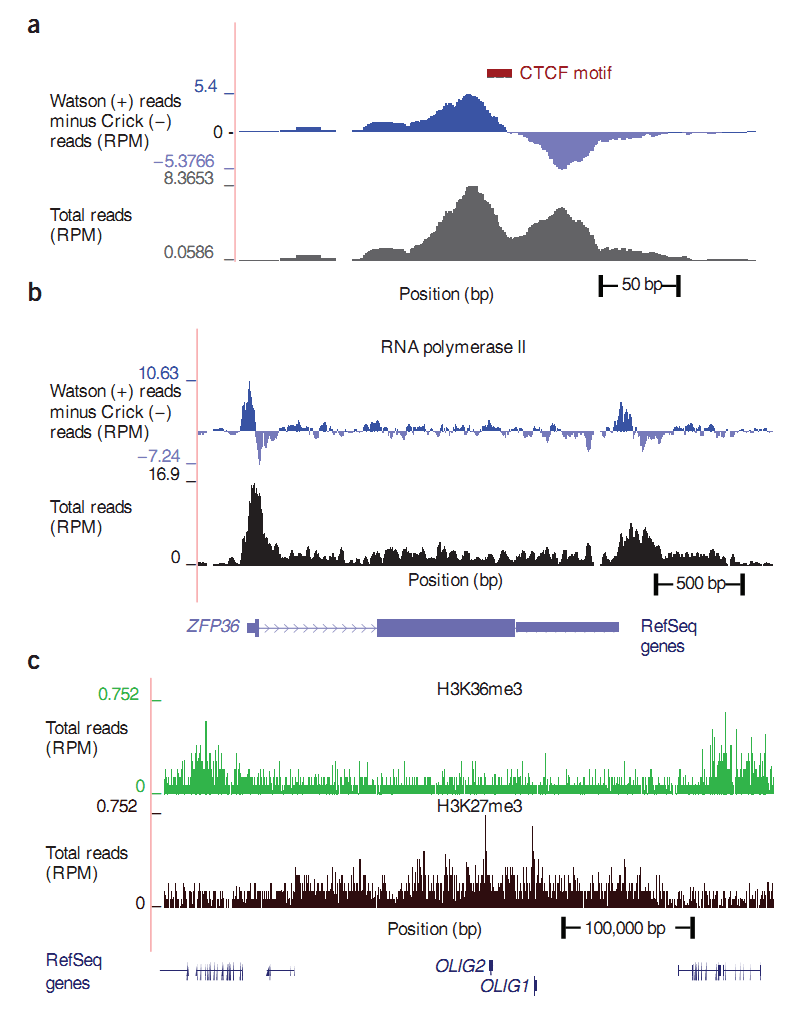 Fig. 2
Fig. 2The presence of the background forces the algorithms to try to evaluate it empirically based on the “control”. The control is an additional experiment carried out with the initial solution with special (preimmune) antibodies, with which nothing should be connected, but still randomly connected. As a result of this connection, some arbitrary background is obtained. Some algorithms model a possible background, based only on the data, without control. Whichever approach is chosen (with control and without control), the distribution of background reads cannot be unified until the end, since the background level depends on both the type of cells and the tissue from which they are obtained, and the method of sedimentation, etc. . The technology used for amplification (PCR) and sequencing can also add artificial enrichments (artifacts) or enhance one region more than another. There are algorithms that allow you to find enriched areas for each experiment. Although each of the algorithms is tailored to the data corresponding to the experiment, the assumption underlying the algorithm does not always fit the possible set of enriched areas found using DNA-seq [2].
Below, for comparison of data, graphs from a more recent article on ZIMBA. Similar graphs are blurred due to the difference in scale between the first and second pictures, but they look similar to the above explanation about the DNA-seq technology (see Figure 2).

Fig. 3
In Figure 3, in addition to the previous graphs, the landscapes of the following DNA-seq experiments are displayed : DNA-sensitive sample (DNase-seq) [5] and protein isolation / fixation with formaldehyde (FAIRE-seq) [6].
DNA is a double helix. This phrase itself is popular, but very often it is forgotten. What does it mean in our case? And the fact that the program that will search for the reads on the genome will give out, along with the coordinates on the chromosome, and from which “side” of the helix (strand) this read is located. Spirals are divided into positive (sense) and nonpositive (nonsense / antisense). The mechanism for obtaining reads is such that most reads will be located on two different sides with respect to the protein bound to DNA. And in most cases, the reads will be located from the 5 'end of the corresponding helix. Therefore, reads from both sides will tend to the center of the protein binding site. Figure 4 depicts this schematically, red dots are reads from the 5 'end, and in the center is a protein bound to DNA.
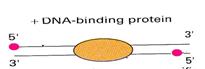
Fig. four
Thus, to find the center of the protein binding site with DNA, we construct the following graph. To do this, we postpone the difference between the number of reads from the positive and negative sides along the “y” axis, and postpone the read coordinate corresponding to the chromosome along the “x” axis. In Figure 5, reads are displayed on the positive side in red, and on the negative side - in blue. The resulting graph crosses zero very close to the center of the protein binding site (see the lower graph in Fig. 5). There are algorithms that, up to a nucleotide, determine binding sites.
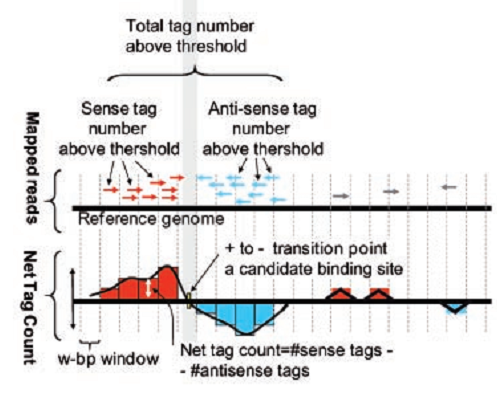
Fig. five
In this topic, I tried to briefly introduce the material that is the object of study. The differences in the landscapes of the corresponding DNA-seq experiments were shown, and the method of approximate finding the center of the protein binding site was explained. There is a lot of work ahead: we need to understand the differences between the algorithms depending on the landscape, learn how to find peaks and try to distinguish them from artificial ones, find out which regions of the genome lying in the vicinity of the peak can be considered enriched, and all this in order to start thinking about ZINBA .
1. Birney, E., et al., Identification and analysis of the functional elements in the 1% of the human genome by the ENCODE pilot project. Nature, 2007. 447 (7146): p. 799-816.
2. Pepke, S., B. Wold, and A. Mortazavi, Computation for ChIP-seq and RNA-seq studies. Nat Methods, 2009. 6 (11 Suppl): p. S22-32.
3. Barski, A. and K. Zhao, Genomic location analysis by ChIP-Seq. J Cell Biochem, 2009. 107 (1): p. 11-8.
4. Barski, A., et al., High-resolution profiling of the human genome. Cell, 2007. 129 (4): p. 823-37.
5. Boyle, AP and TS Furey, High-resolution mapping studies and gene regulatory elements. Epigenomics, 2009. 1 (2): p. 319-329.
6. Giresi, PG and JD Lieb, Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods, 2009. 48 (3): p. 233-9.
Review by Andrey Kartashov, Cincinnati, OH, porter@porter.st
corrected by Ekaterina Morozova, ekaterina@porter.st.
Source: https://habr.com/ru/post/137453/
All Articles