Practical bioinformatics, part 2
This article will tell you how to process the data obtained after the pipeline, the output of which will be the sam / bam file [1], create a simple bed graph file (http://genome.ucsc.edu/FAQ/FAQformat.html) and view it using the UCSC genome browser [2]. It is very difficult to decide on what to write programs for, because there are already a huge number of foreign developments and I don’t want to write a wheel at all where this stage has been passed. Long suffering, I decided to stop at C ++, although Python and R were treated as equals. There is also an idea that graphics may be needed, and even under Linux, so Qt has been added to C ++. I hope that in this article I will tell you in sufficient detail about all of the above to answer the question asked at the beginning of my journey and voiced in the first part of the story.
To work with sam / bam files, we need the samtools [1] package compiled. Download the package from the site samtools.sourceforge.net , expand to a directory, for example, / usr / src go to the created directory and type make. I did not install XCurses on my system and I replaced the line “LIBCURSES = -lXCurses” with “LIBCURSES = -lncurses” and everything was assembled. The result of the make program is the compiled program samtools and the library libbam.a.
We need C ++ classes that will store information about the reads, genes, introns, exons, etc. For the organization of such classes, I used boost.intervals, although not quite in the form that I wanted. Neither boost.intervals, nor boost.icl allow storing complete information about segments. I need the following information about the set of segments:
In particular, this information will answer the question what is the height of the coverage with segments of the current segment of the coordinate axis.
With each new article I will replenish and change these classes. Now they are enough to build a program and estimate what is missing for the future. Here is an example of the resulting classes.
Reads.hpp
Now let's get down to wrapping functions for working with sam files. The difference between sam and bam is that the first is a plain text file, albeit formatted, and the second is compressed and binary, i.e. structured. I prefer to work with bam with a large number of reads, the sam file can reach gigabytes.
BEDHandler.hpp
')
BEDHandler.cpp
I will not give the rest of the code in the article; here is the link to the archive , the structure is quite simple: at the root are two directories, thirdparty and src, in the first one there is samtools, in the second one - sam2bedgraph and global. In order to build a project, you need to run qmake and then make in the sam2bedgraph directory. Checked under openSUSE 12.1 x64 with native Qt (4.7.4) and boost (1.46.1).
The link to the description of the structure of the bedgraph file is given in the first paragraph, but I will briefly mention it. The first line specifies the characteristics of the file, if present. The file body contains at least 4 columns. The first column indicates the name of the chromosome, in the second and third - the coordinates of the beginning and end of the segment ("window"), respectively. In the last column - “height”, in our case, this number began reading on this window. The size of the "window" for each file is best chosen experimentally. I use 200 bp for ChIP-seq, 20 bp for RNA-seq.
A program can have two parameters: an input .bam file and an output file where the bedgraph data will be written. Parameters can be not set, then the program will try to open the default file input.bam and create output output.data. In the user's home directory, a directory will be created with the configuration file .config / CCHMC / sam2bedgraph.ini, in which you can change the default file name values, change the length of the “window” for which we count heights and set the appearance of the first information line.
The resulting file can be downloaded to the genome browser. Go to the site genome.ucsc.edu [2] click on the link Genomes.
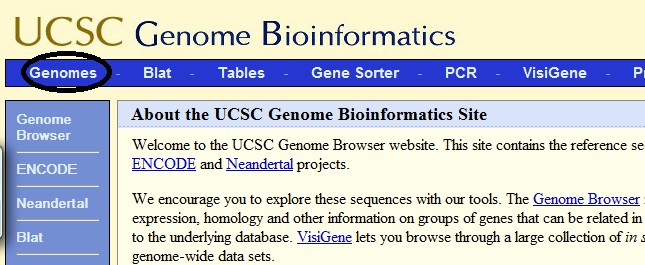
We get to the next page, where it is necessary to set the conditions under which our bam file was received: Clade (Evolutionary Line), genome (Genome), assembly (Abstract, Assembly). And then click the “add custom tracks” button.
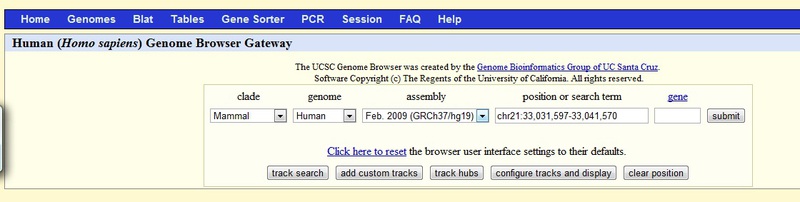
Now you can click the “Choose file” button and upload our file by clicking the “Submit” button. As a result, we get to the page with the annotation, which will be displayed information from the database and from our bedgraph. If you install the genome browser locally, you can add a bedgraph to the site not temporarily during the session, but constantly. You can organize special links to the site, which will indicate where to get the bedgraph file. The site contains instructions on how to copy the genome browser.
You can upload multiple files, which I did. You can see the result in the following screenshot.
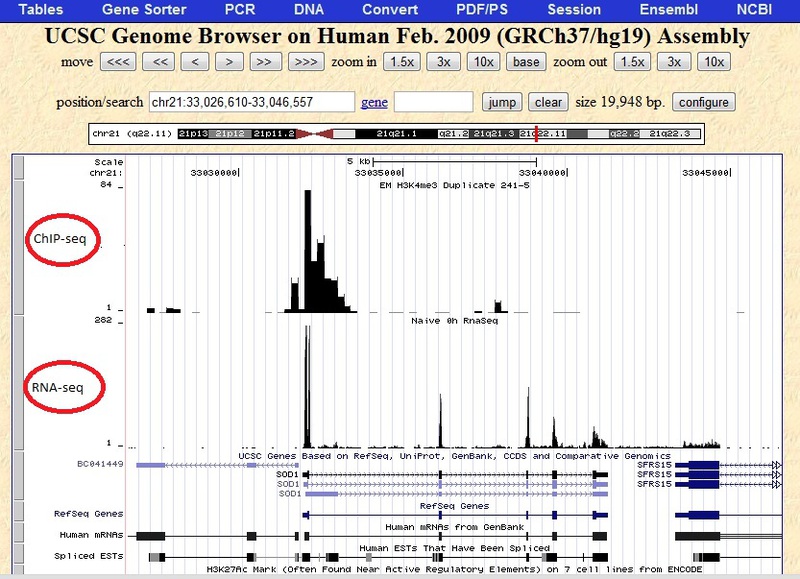
As can be seen in this screenshot, there are areas with a zero level, and there are areas with enrichment, where there are peaks. Sometimes there is a background or noise. You can also clearly see the difference between ChIP-seq and RNA-seq. The definition of these sections from the program is a separate question, the unequivocal answer to which is not. The fact is that there can be as many peaks as there are genes in the genome or, worse, as exons. And to separate the enriched areas in the experiment is very time consuming.
1. Li, H., et al., The Sequence Alignment / Map format and SAMtools. Bioinformatics, 2009. 25 (16): p. 2078-9.
2. Kent, WJ, et al., The human genome browser at UCSC. Genome Res, 2002. 12 (6): p. 996-1006.
3. Dreszer, TR, et al., The UCSC Genome Browser database: extens and updates 2011. Nucleic Acids Res, 2012. 40 (Database issue): p. D918-23.
To work with sam / bam files, we need the samtools [1] package compiled. Download the package from the site samtools.sourceforge.net , expand to a directory, for example, / usr / src go to the created directory and type make. I did not install XCurses on my system and I replaced the line “LIBCURSES = -lXCurses” with “LIBCURSES = -lncurses” and everything was assembled. The result of the make program is the compiled program samtools and the library libbam.a.
We need C ++ classes that will store information about the reads, genes, introns, exons, etc. For the organization of such classes, I used boost.intervals, although not quite in the form that I wanted. Neither boost.intervals, nor boost.icl allow storing complete information about segments. I need the following information about the set of segments:
- any pair of start / end, start / length;
- all segments are closed, i.e. include ends;
- how many segments begin at a given point;
- how many segments intersect at a point;
- how many segments start in a given segment or intersect with a given segment.
In particular, this information will answer the question what is the height of the coverage with segments of the current segment of the coordinate axis.
With each new article I will replenish and change these classes. Now they are enough to build a program and estimate what is missing for the future. Here is an example of the resulting classes.
Reads.hpp
#ifndef _READS_29122011_HPP_ #define _READS_29122011_HPP_ #include <boost/numeric/interval.hpp> #include <QString> #include <QMap> #include <QtDebug> namespace genome { namespace bni = boost::numeric; typedef bni::interval<int> read_position; /********************************************************************************** **********************************************************************************/ class Read { public: Read(): multiplying(1), length(0){}; Read(Read const &r): multiplying(r.multiplying), length(r.length), position(r.position){ sentenceRepresentation=r.sentenceRepresentation; qualityRepresentation=r.qualityRepresentation;}; Read(int start,int len,QString sr="",QString qr=""): multiplying(1), length(len), position(start,start+len-1), sentenceRepresentation(sr), qualityRepresentation(qr){}; int getLevel() {return multiplying;}; void plusLevel() {++multiplying;}; int getStart() {return position.lower();}; int getLength() {return length;}; void operator+= (const int& c) {this->multiplying+=c;}; bool operator== (const Read& r) const {return this->position==r.position;}; bool operator!= (const Read& r) const {return this->position!=r.position;}; void operator++ (int) {this->multiplying++;}; private: int multiplying; int length; read_position position; QString sentenceRepresentation; QString qualityRepresentation; }; /********************************************************************************** **********************************************************************************/ typedef QMap<int,Read> cover_map; class Cover { public: Cover():max_len(0){}; void add(Read&); int getHeight(int); int getHeight(int,int); int getStarts(int); int getStarts(int,int); QList<int> getStarts(); cover_map::iterator getBeginIterator(){return covering.begin();}; cover_map::iterator getEndIterator(){return covering.end();}; bool operator== (const Cover& c) const {return this==&c;}; bool isEmpty(){return covering.size()==0;}; // static Cover empty(){ return Cover();}; private: cover_map covering; int max_len; }; /********************************************************************************** **********************************************************************************/ class Lines { public: Lines(){}; Lines(Lines&){}; void addLine(QString, Read&); Cover& getLineCover(QString); QList<QString> getLines(void) { return lines.keys(); }; /* */ private: QMap<QString,Cover> lines; }; /********************************************************************************** **********************************************************************************/ class GenomeDescription: public Lines { public: quint64 notAligned; // number of reads (ussualy form sam/bam file) that are not aligned quint64 total; /* */ void setGene(const QChar &sense,const QString &chrName,const qint32 &pos,const qint32 &,const qint32 &len) { Read r(pos,len); addLine(chrName+sense,r); }; /* */ GenomeDescription():Lines(), notAligned(0), total(0) {}; }; } #endif Now let's get down to wrapping functions for working with sam files. The difference between sam and bam is that the first is a plain text file, albeit formatted, and the second is compressed and binary, i.e. structured. I prefer to work with bam with a large number of reads, the sam file can reach gigabytes.
BEDHandler.hpp
')
#ifndef BEDHandler_H #define BEDHandler_H #include <config.hpp> #include <Reads.hpp> template <class Storage, class Result> class BEDHandler : public QState { private: Storage *sam_input; Result *output; QSettings setup; QFile _outFile; public: BEDHandler(Storage &sam,Result &output,QState *parent=0); ~BEDHandler(); protected: virtual void onEntry(QEvent* event); }; #include <BEDHandler.cpp> #endif BEDHandler.cpp
//------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> BEDHandler<Storage,Result>::~BEDHandler() { } //------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> BEDHandler<Storage,Result>::BEDHandler(Storage& sam,Result &_output,QState * parent): QState(parent), sam_input(&sam), output(&_output) { _outFile.setFileName(setup.value("outFileName").toString()); _outFile.open(QIODevice::WriteOnly|QIODevice::Truncate); } //------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> void BEDHandler<Storage,Result>::onEntry(QEvent*) { if(!setup.contains("graphWindow")) setup.setValue("graphWindow",200); if(!setup.contains("siteShift")) setup.setValue("siteShift",75); if(!setup.contains("separateStrand")) setup.setValue("separateStrand",false); if(!setup.contains("HeaderString")) setup.setValue("HeaderString","track type=bedGraph name=%1"); if(setup.value("HeaderString").toString()!="") { _outFile.write((setup.value("HeaderString").toString().arg(_outFile.fileName())+"\n").toLocal8Bit()); _outFile.flush(); } quint32 window=setup.value("graphWindow").toUInt(); quint32 shift= setup.value("siteShift").toUInt(); QString line; QString chrome; foreach(line,sam_input->getLines()) { if(line.endsWith("-")) continue; chrome=line; chrome.truncate(line.length()-1); QMap <int,int> bed; { genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('+')).getBeginIterator(); genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('+')).getEndIterator(); while(i!=e) { int val=i.key()+shift; bed[val-val%window]+=i.value().getLevel(); ++i; } } { genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('-')).getBeginIterator(); genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('-')).getEndIterator(); while(i!=e) { int val=i.key()-shift; if(val<0) val=0; bed[val-val%window]+=i.value().getLevel(); ++i; } } QMap<int,int>::iterator i = bed.begin(); for(;i!=bed.end();i++) { _outFile.write(QString(chrome+"\t%1\t%2\t%3\n"). arg(i.key()).arg(i.key()+window).arg(i.value()).toLocal8Bit()); _outFile.flush(); } } }//end of function I will not give the rest of the code in the article; here is the link to the archive , the structure is quite simple: at the root are two directories, thirdparty and src, in the first one there is samtools, in the second one - sam2bedgraph and global. In order to build a project, you need to run qmake and then make in the sam2bedgraph directory. Checked under openSUSE 12.1 x64 with native Qt (4.7.4) and boost (1.46.1).
The link to the description of the structure of the bedgraph file is given in the first paragraph, but I will briefly mention it. The first line specifies the characteristics of the file, if present. The file body contains at least 4 columns. The first column indicates the name of the chromosome, in the second and third - the coordinates of the beginning and end of the segment ("window"), respectively. In the last column - “height”, in our case, this number began reading on this window. The size of the "window" for each file is best chosen experimentally. I use 200 bp for ChIP-seq, 20 bp for RNA-seq.
A program can have two parameters: an input .bam file and an output file where the bedgraph data will be written. Parameters can be not set, then the program will try to open the default file input.bam and create output output.data. In the user's home directory, a directory will be created with the configuration file .config / CCHMC / sam2bedgraph.ini, in which you can change the default file name values, change the length of the “window” for which we count heights and set the appearance of the first information line.
The resulting file can be downloaded to the genome browser. Go to the site genome.ucsc.edu [2] click on the link Genomes.
We get to the next page, where it is necessary to set the conditions under which our bam file was received: Clade (Evolutionary Line), genome (Genome), assembly (Abstract, Assembly). And then click the “add custom tracks” button.
Now you can click the “Choose file” button and upload our file by clicking the “Submit” button. As a result, we get to the page with the annotation, which will be displayed information from the database and from our bedgraph. If you install the genome browser locally, you can add a bedgraph to the site not temporarily during the session, but constantly. You can organize special links to the site, which will indicate where to get the bedgraph file. The site contains instructions on how to copy the genome browser.
You can upload multiple files, which I did. You can see the result in the following screenshot.
As can be seen in this screenshot, there are areas with a zero level, and there are areas with enrichment, where there are peaks. Sometimes there is a background or noise. You can also clearly see the difference between ChIP-seq and RNA-seq. The definition of these sections from the program is a separate question, the unequivocal answer to which is not. The fact is that there can be as many peaks as there are genes in the genome or, worse, as exons. And to separate the enriched areas in the experiment is very time consuming.
1. Li, H., et al., The Sequence Alignment / Map format and SAMtools. Bioinformatics, 2009. 25 (16): p. 2078-9.
2. Kent, WJ, et al., The human genome browser at UCSC. Genome Res, 2002. 12 (6): p. 996-1006.
3. Dreszer, TR, et al., The UCSC Genome Browser database: extens and updates 2011. Nucleic Acids Res, 2012. 40 (Database issue): p. D918-23.
Source: https://habr.com/ru/post/137082/
All Articles